
Analyzing gel electrophoresis results and interpreting them, is a bit difficult task. One has to develop skills to read a gel. Let’s explore how you can do that with exclusively real gel examples.
Gel electrophoresis is a conventional, native and subsidiary technique used to visualize DNA. There are two types of gel electrophoresis but we use conventional agarose gel electrophoresis so often.
It requires an agarose polysaccharide that forms a porous structure for DNA to migrate. Using a colored marker the migration can be monitored and reported once the run is completed.
Usually, Bromophenol blue is a dye used to track the migration, while EtBr is used to visualize DNA under UV light. The present technique is simple, handy and super useful, but still, requires a lot of experience.
One has to have the expertise to perform, study, read and interpret the results. Often times students find it difficult to read gel results due to the lack of literature regarding this topic.
So I planned to write this article.
I have years of experience working in molecular laboratories. Using the best of my knowledge and expertise I will explain how you can read gel electrophoresis results. This article contains real gel images, some good and some bad.
Actually, I especially collected such bad gel plates so that I can make you understand how good and bad things look in a gel. In the journey, first I will let you know how you can read the gel electrophoresis results.
So this article would be a fun thing and a learning assignment for you and surely help you in your PCR and gel electrophoresis venture.
Stay tuned.
Key Topics:
How to read gel electrophoresis results?
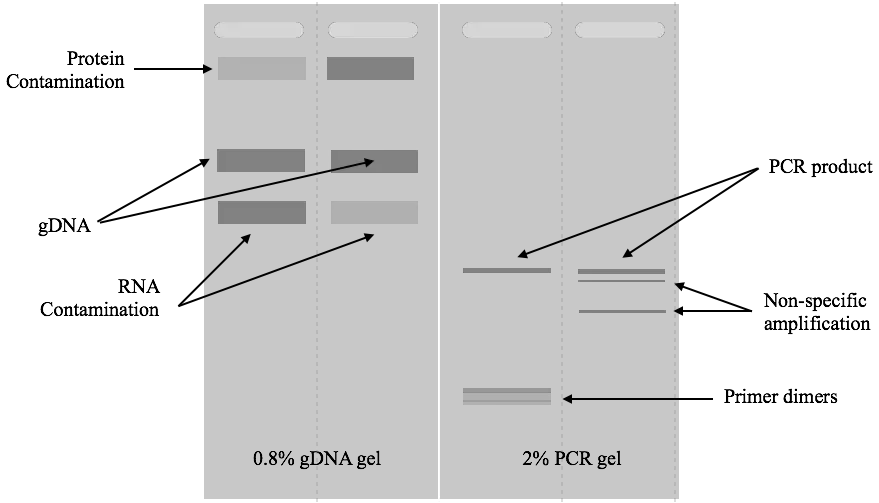
First, make clear if a gel contains any results or not. For that, put the gel carefully under the UV light and see if it contains any bands or not.
In the second step, see if the gel possesses any visible contaminants like protein or RNA, or not. Contaminants have a direct effect on the purity of DNA and hence we have to identify them next.
If the gel has an RNA contaminant, the RNA bands will appear above the DNA. RNAs are smaller than DNA and thus migrate faster. So thick or many smear-like bands of RNA will appear above the DNA band which indicates RNA contamination.
Likewise, if any thick band appears below the DNA, it will indicate protein contamination. The protein migrates behind the DNA in the gel.
If any things mentioned here doesn’t appear in the gel, move ahead to investigate primer dimers. Primer dimer, in the case of PCR results, is an important marker that shows if the amplification occurred correctly or not. Here, some complementary nucleotides of both primers bind and amplify in the reaction.
Such products are short, usually 20 to 50 bp and appear at the bottom of the gel, far away from the DNA. If you see any faded band there, make sure you have primer dimers in the reaction.
A thick band of genomic DNA, a linear and sharp band of PCR and a very sharp band of restriction digestion will appear in the gel. This is how you can read gel results. Take a look at the images below.
Related article: Factor Affecting DNA Agarose Gel Electrophoresis Results.
Now take a look at some of the real gel pictures and discuss them one by one.
Gel electrophoresis results of gDNA:
Image 1

This image is non-conclusive actually. But if you carefully observe well 9, the DNA is trying to come out from the gel. However, the smear indicates the contamination of RNA and DNA degradation.
Image 2

From 64 to 79, in each well DNA is trying to come out of the well but some DNA remained inside the well. A couple of reasons are responsible for that Firstly, the wells are broken during sample loading (see 72, 74, 75, 76, 77, 78) and secondly, the air bubbles were formed during the gel casting.
One another possibility in this gel is that the comb is not placed correctly or the gel is disturbed during the removal of the comb. Because of these reasons, the gDNA is unable to come out from the well.
Moreover, the smears above and below the DNA indicate contamination of RNA and protein. See wells 75, 76, 77, 78, 79. Conclusively, the DNA is not extracted properly and the gel preparation is poor.
Image 3

This image is in two parts, part one, wells 48-55 is non-conclusive while part two from wells 56-63 shows some abnormalities. The best concentration of DNA is in the well 59, although due to poor gel preparation, it can’t come out properly.
From wells 56 to 63, the gel wells are disrupted and also the DNA is contaminated with protein and RNA.
Image 4

Image 4 shows DNA degradation, contamination with RNA and proteins, poor gel preparations and unhealthy lab practices. Although, a substantially good amount of DNA is obtained in wells 45 and 47 but is of poor quality.
Common problems in this gel are improper comb setup, poor gel quality, disrupted wells, etc.
Image 5

Now, this case is a bit different from other gels. Here the gel loading buffer is reused so many times, therefore, the actual concentration of the buffer is changed during the electrophoresis of this gel. Due to this reason, the buffer limits the migration of DNA and therefore smears of DNA bands appeared.
Also, the gel is slightly brighter than other gels because of the presence of fragments of other previous DNA (in each run some amount of DNA remains in the buffer which appears in the next run when we re-use it).
Image 6:

This is a decent image of human blood genomic DNA extraction. You can see how intact the bands are and how beautiful it looks. This is an image of gDNA on 0.8% agarose gel. I also have added two other plates showing substantial RNA contamination in the gel.
Related article: What is Electropherogram? How to Read it?
Gel electrophoresis results of PCR:
The results of PCR are run on 2% gel with a clear and known DNA ladder. Now take a look at some of the results of PCR.
Image 1:
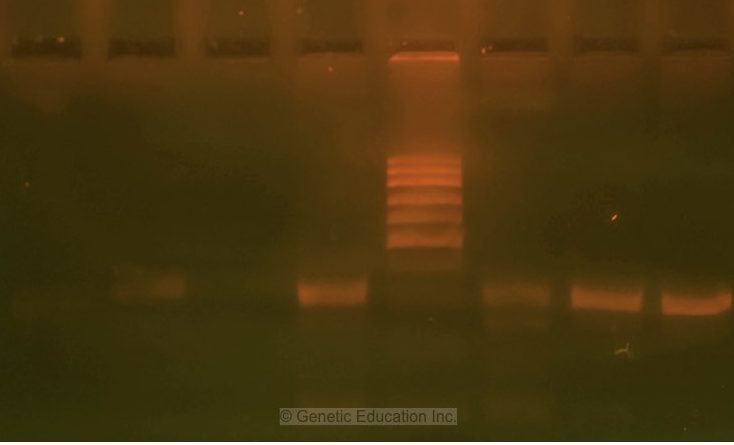
The image is captured under the UV transilluminator instead of the gel doc system to show you the effect of EtBr on the gel electrophoresis results.
Here due to the re-use of a gel as well as the buffer, the EtBr is not properly spread into the gel. Further, the traces of the previous EtBr is also present in the gel. See the orange color near the wells, DNA and ladder. These all are the EtBr molecules not spread well.
Due to poor gel preparation, the ladder, as well as DNA, are not separated well.
Image 2:

This image is very special, we have re-used gel and buffer many times. You can see the condition of DNA bands. Results are non-conclusive and fresh new electrophoresis gel and buffer are required here.
Image 3:
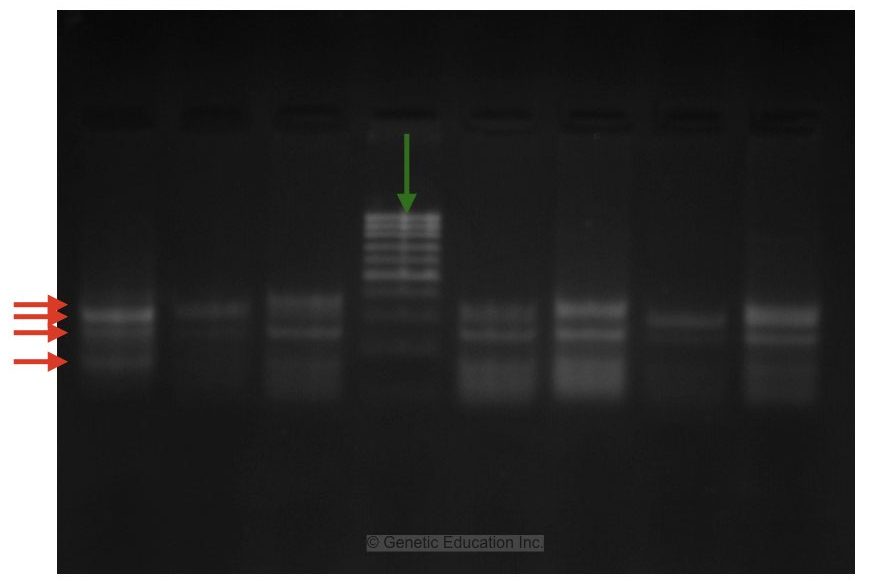
Now this image is pretty good but what is the problem? You can see many bands, right?
Here the annealing temperature of the primer is not selected properly. So the primer is compromised with other complementary sequences present in the genome. The annealing temperature is too low in comparison with its actual annealing temperature.
Due to this reason, more than 4 bands of PCR amplicons are observed in the gel. I also have noticed a minor thing. See the green arrow, it shows an air bubble in the gel. Conclusively, the PCR is not performed with the optimum PCR conditions.
Image 4:
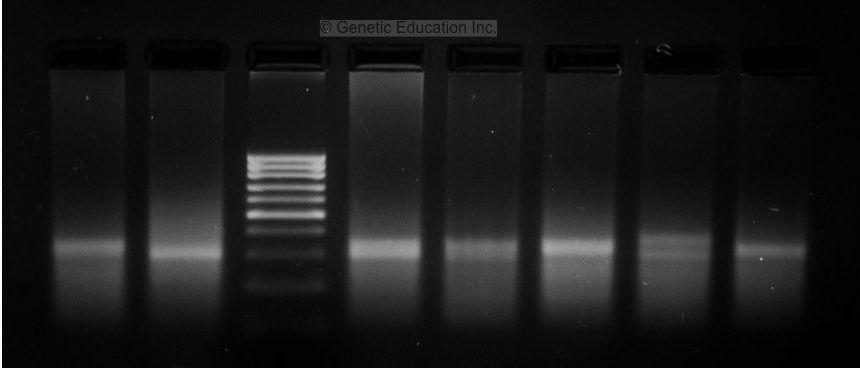
Now this gel is pretty good, isn’t it? The DNA ladder is separated nicely and DNA is also appropriately amplified. But the concentration of the template DNA is a problem here.
The concentration of the template DNA used in this PCR reaction is very high. In a normal PCR reaction, 25 to 30ng concentration is sufficient. However, in this PCR reaction, the concentration of DNA will be more than 100ng.
The smear of the DNA along with the amplified product is observed due to this reason. And also two bands in some reactions, if you can see it.
Image 5:

This gel is in very bad condition, nothing is good here.
The shining dots in the gel are air bubbles. Due to the air bubbles, the ladder is not migrated properly see the first red arrow. Further, the ladder and DNA are too old, or not maintained properly, it’s degraded.
Image 6:
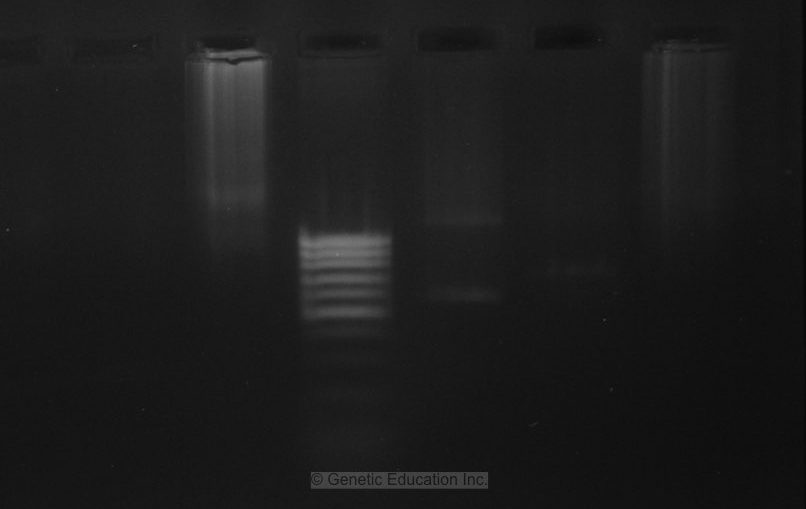
Now analyze this gel image, the DNA ladder ran faster than the samples. The samples are smeared as well which means that the buffer is too old, its concentration is altered or the pH of the buffer may be probably changed.
Remember, when we have the smears like this in any of the PCR products our buffer is the problem.
Image 7:
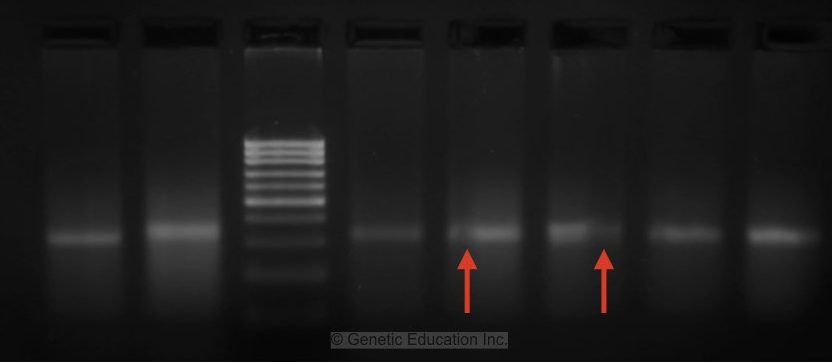
Now this image is pretty good but still has some problems. See the two red arrows, the bubbles which interfere with the migration of the DNA, and the higher concentration of the template DNA.
We have seen all the types of DNA gel electrophoresis results and interpreted each type of electrophoresis results. But what qualities does a good-quality electrophoresis gel has?
- Good and sharp bands
- Minimum primer-dimers
- A beautifully separated DNA ladder.
- No background or traces of other DNA in the gel
See the next gel image and analyze each parameter. Though the primer dimers are present that is another issue. The result of the gel is beautiful and the bands are so clear and self-explanatory.
Image 8:
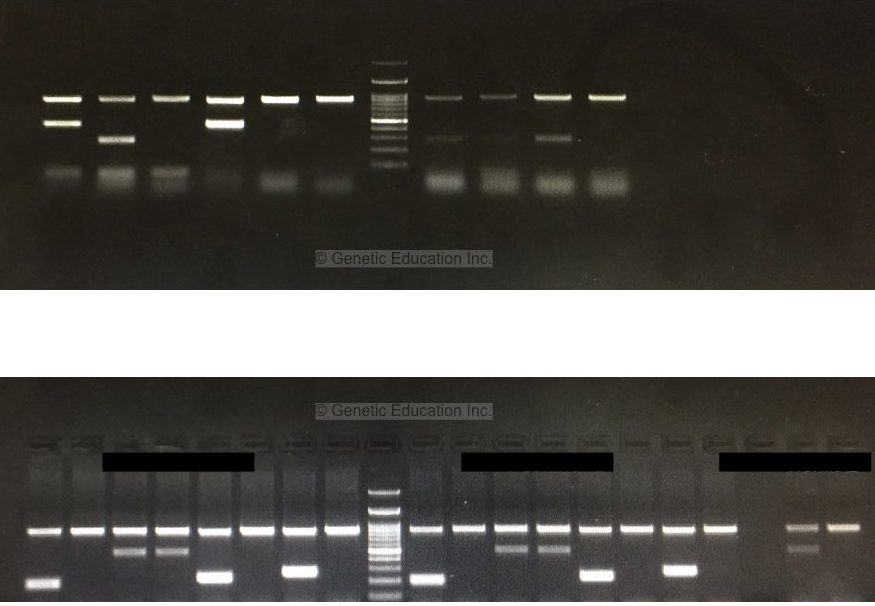
So after all these observations, I have a few suggestions which eventually help you more in this learning.
How to get good gel results?
- Do not re-use the gel. If necessary use only twice.
- Do not reuse the buffer. If necessary use only twice or thrice.
- Prepare buffer freshly every time for the gel as well as the electrophoresis tank.
- Preserve DNA and DNA ladders properly in the cold chain.
- Use template DNA ~30ng to 50 ng not more than that, in the PCR reaction.
- Use only 10pMol primers. Follow PCR protocol.
- Use high-quality chemicals.
Read our next article in this series. The article is on the gel electrophoresis analysis of restriction digestion, cccDNA, linear DNA, supercoiled DNA and multiplex PCR: Part 2: Analysing and Interpreting (Agarose) Gel Electrophoresis Results.
Wrapping up:
Getting good quality gel electrophoresis results is a matter of expertise. As you do it you will get mastery over time. Nonetheless, to sharpen your skills perform every step precisely.
Gel electrophoresis is an important genetic technique. It is used to validate the results of genomic DNA, PCR amplicons, restriction digestion and DNA library screening. I hope this article will help you in your genetics learning.

Subscribe to our weekly newsletter for the latest blogs, articles and updates, and never miss the latest product or an exclusive offer.

first of all thank you for this wonderful explanation. I want to ask a another question. what could be the reason of brightness in the UV absorbtion images?
Dna visualization uv rays cause disease so another technique use ????
No, we can only visualize DNA into UV with safety precautions.
My question is that in analysing and interpreting of the genomic DNA like in image 3 well 48,49,57 and 58?
We had loaded gDNA in those wells as well but it can’t be extracted or loaded correctly. That is why bands are not seen.
We mean the whole gel is of gDNA samples. Some are contaminated, some are not migrated well, some are not extracted well etc.
hope you understand.
Thank you it’s really helpfull
hello sir, your articles are so helpful and easy to understand. i am masters student and i always face problem in calculation part like if we are provided with this much of stock solution and have to make some amount of working from it. if you could please discuss about that it would be very kind of you.
Sure no prob. Give me tour email i will send you some material
[email protected] thankyou sir for your prompt response.
[email protected]
sir could you please also discuss about protein purification methods like SDS PAGE in future.
Hello Naidu. Our team is enthusiastic to write new things but unfortunately our blog niche is specific to DNA and Genetics so we cant discuss protein purification.
But we will try to cover SDS PAGE.
good source of info
Thank You Ian Bremen
Very useful information
thank you
Very nice and helpful article. Thank you!!
thank you so much for appreciation
Hi, I had prepared a gel electrophoresis result for my dissertation. My lecturer had suggested that I comment about the level of expression of the mRNA. was wondering, what does she mean by this? How can we comment on level of expression of the mRNA?
Even i don’t understand your question? Can you reconferm your question?
Wow i really enjoy the content of this work.May God give you strength to do more.stay Blessed
Thank you so much Augustine
Thank you microbiology online notes. Appreciation from the giant platforms like you is a kind of achievement for us.
very nice content about electrophoresis easly understable keep doing like this..
WOW I REALLY ENJOY THE CONTENT OF THIS WORK IT’S WONDERFUL .MAY GOD GIVE YOU STRENGTH TO DO MORE.STAY BLESSED