“Paired-end sequencing can sequence the fragment from both ends. In this article, learn about the concept, applications and advantages of paired-end sequencing.
Illumina is a pioneer and industry leader in DNA sequencing and NGS platforms. Their technology, unique sequencing chemistries and constant optimizations in the existing technology help improve quality and throughput.
Next-generation sequencing revolutionized the genetic testing and research field by allowing accurate and efficient sequencing of the whole genome. Single-end sequencing, though, is an amazing optimization but has several shortcomings.
It can not effectively sequence longer fragments and repetitive genomic regions. Thus, overall sequencing quality decreases. A recent advancement in the NGS with paired-end sequencing helps overcome these limitations.
Let’s find out how paired-end sequencing works, how it is done and what are its applications. Stay tuned.
Related article: DNA Sequencing: History, Steps, Methods, Applications and Limitations.
Disclaimer: The content presented herein has been compiled from reputable, peer-reviewed sources and is presented in an easy-to-understand manner for better comprehension. A comprehensive list of sources is provided after the article for reference.
Key Topics:
What is Paired-End sequencing?
Paired-end sequencing, as the name suggests, can sequence from both ends of the target DNA sequence. It’s available with the Illumina NGS platforms. It increases the accuracy and efficiency of reads, particularly for repetitive regions.
Paired-end sequencing is available for both DNA and RNA sequencing and has been utilized for genomics and gene expression studies.
How does it work?
So the process begins with the usual nucleic acid isolation, either DNA or RNA, depending on the assay requirement. The isolate is purified, quantified and sent for fragmentation.
In the next step, the nucleic acid is fragmented into smaller DNA fragments. This again depends on the requirement of the assay. Usually, it’s fragmented into 300 bp fragments for 150 bp or less paired-end sequencing.
After that, the fragments are ligated with known sequence adaptors and enriched with amplification. Now, the sequence library is ready for paired-end sequencing.
To use it in the sequencing, along with adapters, the flow cell oligonucleotide complementary sequences and index sequences are added to the template sequence. Usually, the adaptors work as a sequencing primer.
First, see the image before moving head, so that you can better understand the concept.
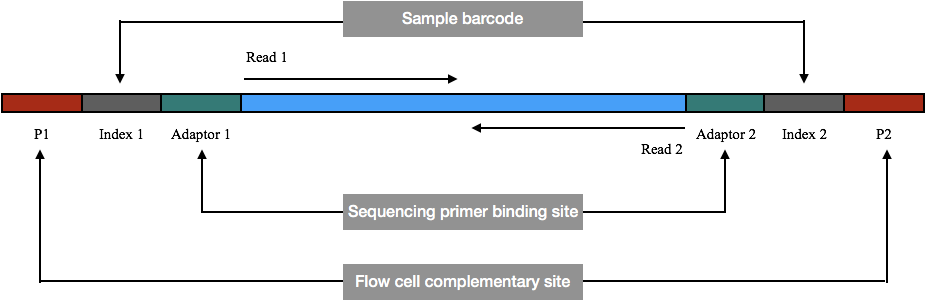
The flow cell is a microscopic slide containing two types of oligonucleotide sequences. One is complementary to P1 and another is complementary to P2 . When we apply the fragmented sample, Each flow cell’s complementary sequences (P1 and P2) are hybridized with its complementary sequence on the flow cell.
The unbound sequences and the original template are washed away. Now, the newly synthesized single-stranded DNA again binds its complementary flow cell oligonucleotides and through bridge amplification, a new DNA strand is synthesized.
The process is then repeated many times and occurs simultaneously in millions of DNA sequence clusters. After completion of the bridge amplification, one last time the single-stranded DNA fragments are generated and sequenced by the synthesis process.
Now, here the index sequences are used as the known sequence tag for the computer during the analysis part.
Related article: What is ‘Sequencing Read’ in NGS?
R1 and R2
Students ask on the Internet what is R1 and R2 in the paired-end sequencing. Here the R1 is the read 1, also known as the forward read and is from 5’ to 3’ direction while the R2 is read 2, also known as the reverse read and is from 3’ to 5’ on the other end.
For example, if we have a 300 bp fragment. The 150 bp forward sequencing is read 1 while the 150 bp reverse sequencing from another side is read 2 (because the read length is 150bp). Note that read (R)1 extends from the adaptor 1 while the read (R)2 extends from the adaptor 2.
Other names of R1 and R2 are listed here.
| Read 1 | Read 2 |
| R1 | R2 |
| Pass 1 | Pass 2 |
| Left read | Right read |
| /1 | /2 |
Index 1 and Index 2
Contrary to R1 and R2, index 1 and index 2 are sample barcodes known to the computer program to locate the sequence accurately. Thus, index sequences are often used to know the location of the adaptors, thereby the first and second read initiation sites.
Paired-end sequencing assembling
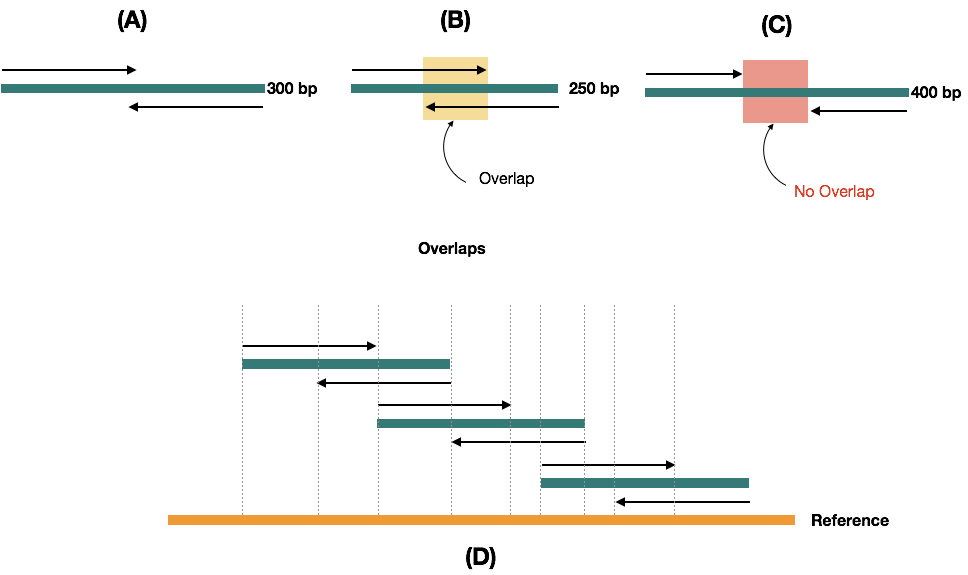
Suppose we have a 300 bp fragment, we can generate two different 150 bp sequence reads, but if we have a smaller fragment than the 300 bp for example 250 bp, overlapping regions are generated.
Contrary, if the fragment is >300bp in site, no overlap is generated. Note that overlapping regions play an important role during the read assembling and accurately locating each nucleotide with the reference sequence. The entire scenario has been explained in (A), (B), (C) and (D) images.
During the sequencing by synthesis process, first read 1 will be sequenced which is up to 150 nucleotides and then read 2 is sequenced which is also up to 150 bp, from the reverse side. So, the same single strand is sequenced from both the ends simultaneously.
Due to this reason, two separate FASTQ files, one for R1 and another for R2 are generated during the paired end sequencing. The R1 FASTQ file has the sequence and overlap information regarding the forward sequencing while the R2 FASTQ file has the sequence information regarding the R2 read.
Why Is It Better?
Let’s discuss the advantages and applications of paired-end sequencing in this section.
Advantages of Paired-end sequencing:
- The present sequencing optimization enables scientists to ‘more’ accurately sequence smaller-sized fragments.
- It’s useful for DNA and RNA sequencing, so, gene and gene expression studies.
- The library preparation and workflow are simple to use and generate high-quality and overlapping reads.
Applications of Paired-end sequencing
- The present assay has been utilized for sequence-based studies and gene expression analysis without the requirements of disulfite treatments.
- It can effectively study the larger and repetitive genomic regions.
- It is used in gene fusion and slice isoform studies.
Related article: 47 Types of Sequencing Techniques You Should Know About.
Wrapping up:
NGS is an interesting and amazing sequencing platform. Although it looks like a complex and tedious thing! But it’s a game of utilizing your expertise- How much you know the platform!
Sequencing by synthesis and paired-end sequencing has revolutionized the RNA-seq and genome analysis field entirely. I hope your concept regarding the paired-end sequencing is now clear.
If you like this article, share it with other researchers.
Sources:
Advantages of paired and single-read sequencing by Illumina.
Liu, T., Chen, CY., Chen-Deng, A. et al. Joining Illumina paired-end reads for classifying phylogenetic marker sequences. BMC Bioinformatics 21, 105 (2020). https://doi.org/10.1186/s12859-020-3445-6.