
“Common problems in gel electrophoresis are the absence of bands, band smears, weak resolution, diffused bands, wavy bands and poor band separation. This article comprises common troubleshooting to overcome these problems.”
Native gel electrophoresis has widespread use in molecular genetic studies for DNA or RNA visualization and analysis. The working principle of gel electrophoresis is the “separation of charged molecules based on their size, shape and charge”.
The present technique is widely used in PCR studies, DNA fingerprinting, restriction digestion, and investigation of mutations and genetic diseases. However, much like PCR, scientists have to encounter several problems during electrophoresis.
I have years of experience working in a genetic laboratory and performed tons of DNA isolation, gel electrophoresis and PCR. In this article, I will explain some of the common problems that occur in gel electrophoresis of DNA or RNA and possible troubleshooting associated.
Stay tuned.
Key Topics:
Common Issues in DNA and RNA Gel Electrophoresis and Troubleshooting
No bands:

The absence of any bands is a common problem newbies face during electrophoresis. When they are doing their first PCR and electrophoresis oftentimes, they get nothing. It may happen with an experienced person too. This problem can be divided into two types, “no any bands (marker + sample both not present) and no bands in sample lanes only (DNA marker is present).
Let’s see each situation one after another.
‘No any bands’ means the DNA ladder (the marker) and sample bands aren’t appearing in the gel. Common reasons for this situation and troubleshooting are explained here.
| Cause | Troubleshooting |
| Absence of tracking dye. The lab personnel forgot to add it to the gel. | Re-strain the gel with EtBr (0.5µG/ml) in the running buffer for at least 20 minutes. Or Re-prepare the gel. |
| Bands are running out of the gel by accidentally wrong electrode placement. | Check if electrodes are correctly placed or not (Red for positive and black for negative). |
| DNA is running out of the gel by the wrong gel placement. | Check if the loading dye is visible or not. |
| The electrodes are placed correctly but accidentally the sample may be electrophoresed off the gel (over-running). | Re-prepare the gel and run the electrophoresis again. Carefully monitor the migration of running dye. |
| Poor or no- amplification can be a reason also but lacks validation as the DNA ladder also does not appear in the gel. | Check your PCR protocol and preparation. |
| The less likely reason is the degradation of the DNA ladder and samples by nuclease action during storage. | Store in appropriate conditions. |
No sample bands:
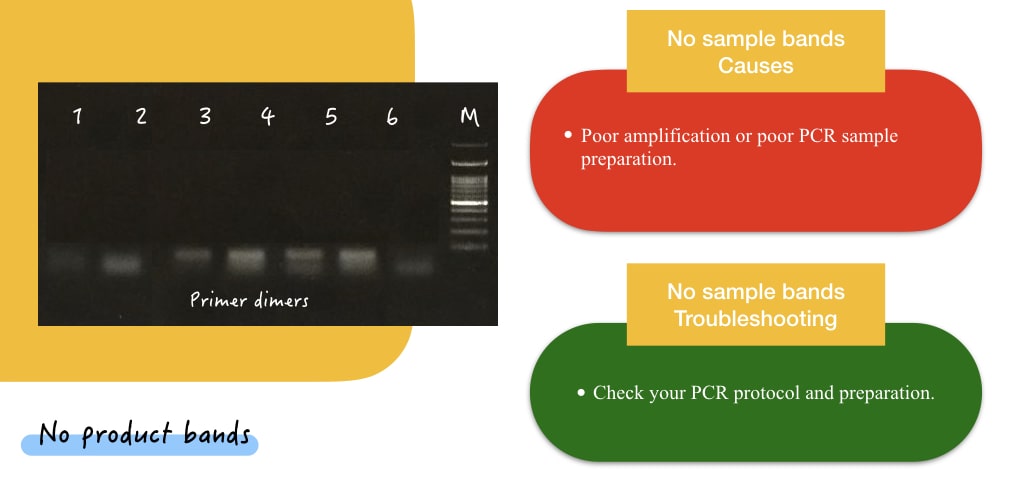
Now the second most common possible result in this category is the no sample bands. Sometimes, you may get fine DNA ladder/marker bands but don’t get bands in any or some sample lanes. It’s a common problem in electrophoresis and can easily be resolved.
| Cause | Troubleshoot |
| Poor amplification or poor PCR sample preparation | Check your PCR protocol and preparation. |
- Here, nothing is wrong with gel electrophoresis. Do check your PCR reaction preparation and protocol. And run again.
Key point: No sample bands are the common problems with PCR amplification and are usually not observed during genomic DNA gel electrophoresis.
Out of the box: No bands indicate inadequate amplification conditions and are caused frequently by incorrect annealing temperature. Decreasing the annealing temperature is one possible solution for PCR.
Dim or weak bands:
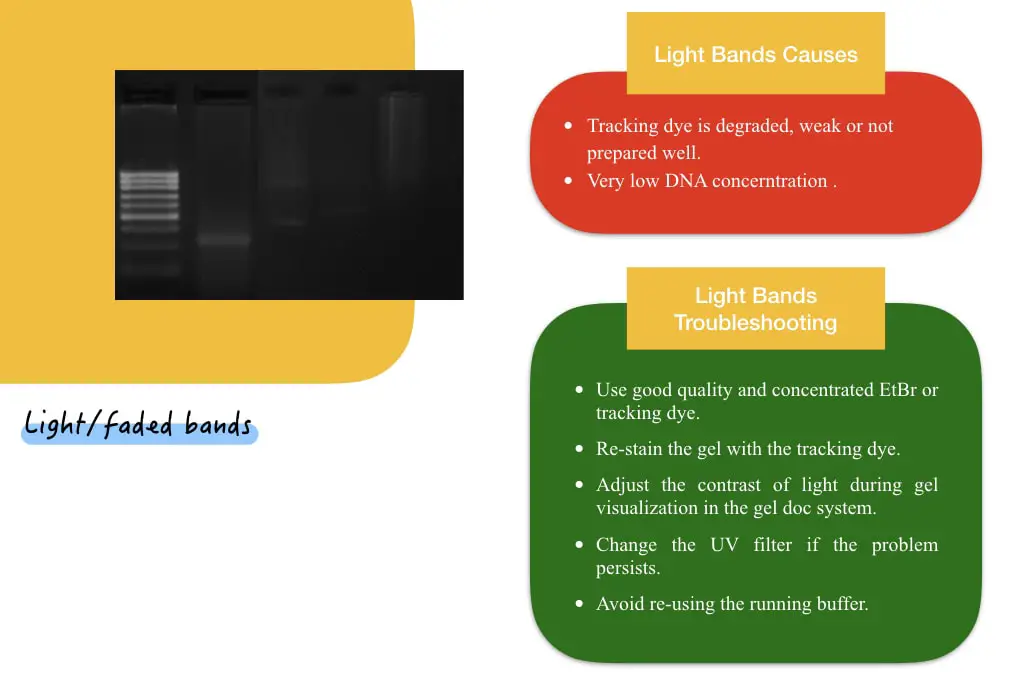
Dim or weak bands lead to poor gel resolution. A DNA/RNA marker and samples or only sample (DNA/RNA) bands are appearing weak or low intensity. This situation can be completely ruled out by understanding two sub-scenario in this category.
If the marker as well as the sample bands both appear very weak, there is something wrong with the tracking dye/EtBr. The stain is weak, low quality or not prepared well.
Possible troubleshooting is here.
- Use good quality and concentration EtBr or any other tracking dye.
- Re-stain the gel with the tracking dye.
- Adjust the contrast of light during gel visualization in the gel doc system.
- Change the UV filter if the problem persists.
- Avoid re-using the running buffer.
- Now if the marker looks decently good but sample bands are dim/weak, the possible reason is low/ weak amplification. Look for your PCR protocol.
Diffused bands:
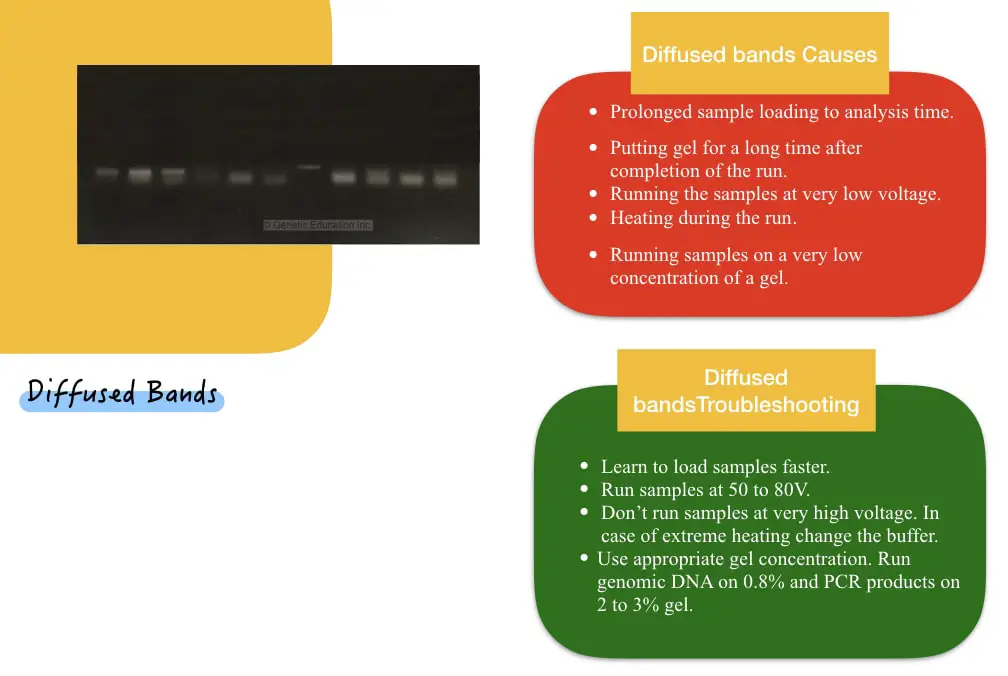
Diffused-looking blurry or fuzzy bands are a serious case of sample diffusion. Meaning, the sample bands are diffused in the surrounding gel pores due to prolonged sample loading or analysis time. Possible causes and relevant troubleshooting are here.
| Cause | Troubleshooting |
| Prolonged sample preparation and analysis time. | Learn to load samples faster. |
| Resting gel for a prolonged time before analysis. | Complete all the analysis preparation before the run ends and analyze samples as soon as possible. |
| Running the samples at very low voltage. | Run samples between 50 to 80V. |
| Heating during the run | Don’t run samples at very high voltage. In case of extreme heating, change the buffer. |
| Running samples on a very low concentration of a gel. | Use appropriate gel concentration. Run genomic DNA on 0.8% and PCR products on 2 to 3% gel |
Smears in the gel:

‘Smears in a gel’ is the most complicated problem to resolve. Many types of smears I have seen with different issues. Tailed smear occurs by higher sample concentration, protein contamination and higher gel concentration.
Headed smear (smear ahead of the sample band) is likely to occur by RNA contamination in the DNA (and vice versa), higher sample concentration and DNA degradation.
In addition, nuclease cleaves DNA or RNA and produces a substantial smear in the gel. Smear in the genomic DNA gel also shows a substantial amount of contamination, higher DNA concentration and poor gel preparation.
Conversely, a smear in the restriction digestion gel which is less likely to occur shows the selection of the wrong restriction enzyme having multiple cutting sites or gel concentration.
| Possible cause | Troubleshooting |
| Higher DNA concentration | Decrease the concentration of DNA and re-run the gel. |
| Tailed and headed DNA smear | Purify the DNA product to remove any contaminant possibly present in the sample. |
| Sample degradation by nuclease action | Store samples in a nuclease-free environment. Take special care of RNA as RNase is present predominantly everywhere. |
| Incorrect gel type | Use adequate gel concentration to run the sample. |
| Incorrect electrophoresis parameter | Don’t run a gel on very high voltage, the temperature of the gel should remain below 30°C and the buffer should have intact buffering capacity. |
| Salt contamination in the DNA | Removed all the salts by alcohol wash before the run. |
| Denatured DNA: If DNA is denatured by extreme heat, it also creates a smear in the gel. | Don’t heat the DNA at extreme temperatures. |
| Fragmented DNA: Accidental fragmentation of DNA by vortexing or nuclease action may also produce a smear in the gel. | Use standard protocols to extract, process and store DNA. |
Other solutions: Don’t reuse the buffer or gel to avoid any smear in the gel.
Band smears may also be produced by incorrect gel preparation, bubbles during gel preparation, or breaking gel during sample loading. This causes samples to be stuck in or near the gel wells. A possible solution for this is to follow healthy gel preparation practices.
Smiley bands:
No wonder, how many times you have seen smiley bands in a gel as a genetic geek. If you think that you have done something different and want to show it off, you are wrong. Smiley bands are common gel electrophoresis anomalies.
A typical characteristic is curved band ends. Smiley bands make interpretation difficult for multiplex PCR results and restriction digestion products, having multiple bands in a single well. Here are common causes and troubleshooting.
| Cause | Troubleshooting |
| Unhealthy gel setups (a comb isn’t placed and removed well, dry wells and wells break). | Follow healthy gel preparation practices. Place and remove the gel smoothly and without any distortion. Ensure to fill each well with a buffer. Do visible observation for gel break or wells anomy. |
| Smiley bands in the entire gel: uneven gel density and older running buffer. | Re-prepare the gel and running buffer. |
Wavy banding pattern:
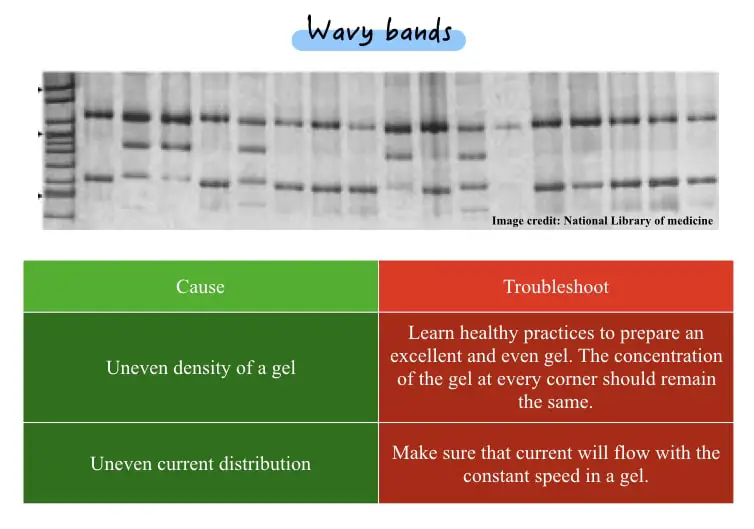
Oftentimes, you may see a wavy banding pattern in your DNA gel. Usually, it is seen during marker-assisted selection gel electrophoresis, like– RAPD or RFLP. Overall, it makes the interpretation part difficult.
| Cause | Troubleshoot |
| Uneven density of a gel | Learn healthy practices to prepare an excellent and even gel. The concentration of the gel at every corner should remain the same. |
| Uneven current distribution | Make sure that current will flow with the constant speed in a gel. |
Note:
Banding anomalies:
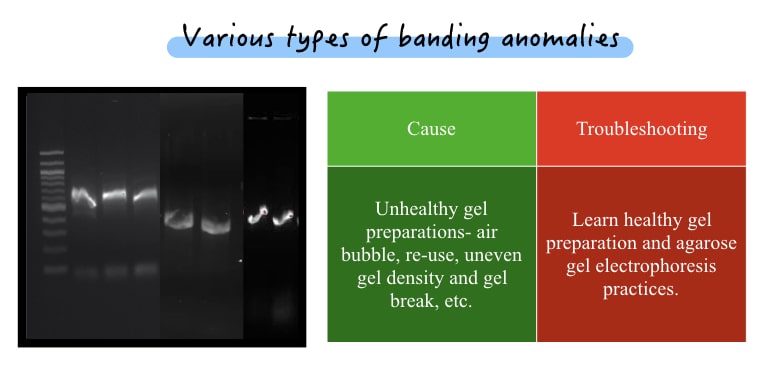
When individual bands aren’t appearing good are categorized into banding anomalies. Common anomalies are breaks in a band, curvy, light, smiley bands and a smear in an individual band. Here are possible solutions.
| Cause | Troubleshooting |
| Unhealthy gel preparations- air bubble, re-use, uneven gel density and gel break, etc. | Learn healthy gel preparation and agarose gel electrophoresis practices. |
Extra bands:
Extra bands appearing in a gel is not a problem associated with gel preparation or electrophoresis, common reasons are associated with PCR, compromised annealing temperature and others. We will discuss it later in the PCR troubleshooting part.
If you see extra bands in your gel, there is something wrong with your PCR setup.
Poor band separation:
Yet another crucial trouble in gel electrophoresis of nucleic acid is poor band separation indicating that bands aren’t separated well from one another. This problem commonly occurred in multiple PCR and restriction digestion.
Poor band separation makes the interpretation part difficult. Let’s check common causes and possible troubleshooting.
| Causes | Troubleshooting |
| Try to run a longer gel to separate smaller product-sized fragments. | Run the electrophoresis completely, until the loading dye reaches the edge of the gel. |
| Slow running | Run electrophoresis at 50 to 80V. |
| Amplified or digested products are too nearer | Try to run longer gel to separate smaller product-sized fragments. |
| Low buffering condition | Oftentimes, decrement in buffering capacity leads to poor band separation. |
DNA remains in the well
The present problem is less likely to occur with PCR products but commonly observed with genomic DNA gel. Some amount of DNA will remain in the gel and create smear-like bands. Here are possible causes and solutions.
| Cause | Select gel concentration depending on the requirement. 2% for PCR and 0.8% for genomic DNA. |
| Gel wells are broken | Place and remove the gel comb correctly. |
| Bubble in wells. | Prepare gel well. Make sure to remove any bubbles near wells. |
| High DNA concentration | Dilute the DNA or decrease the loading amount. |
| Incorrect gelling conditions | Select gel concentration depending upon the requirement. 2% for PCR and 0.8% for genomic DNA. |
Other common DNA/RNA gel electrophoresis problems:
At last, I am also adding some other factors producing unknown anomalies during gel electrophoresis.
- Salt, protein and other contamination in the sample.
- The low density of the loading buffer can’t settle DNA at the bottom of wells.
- Power cut during the run.
- Incorrect running buffer.
- Differences in the buffer used to prepare the gel and run the electrophoresis.
- Uneven staining.
- Incorrect ladder.
- Incompatible running buffer.
- Sample contamination.
- Gel reuse.
All of the above problems can be overcome by following healthy DNA extraction and gel preparation practices.
Wrapping up:
I have limited this article to showing problems and solutions for gel electrophoresis only so that you can understand it. Electrophoresis is a crucial subsidiary genetic technique to study DNA or RNA in genetic research.
However, to work effectively students should know the problems that would occur and possible solutions so that they can optimize their performance. I am hoping this article helps achieve both goals and strength your technical knowledge.
If you like your article, please share it, bookmark the page and buy our ebook, appearing anywhere on this blog.
