“Whole Genome Sequencing is a process of reading an organism’s entire genome. The present student guide explains how the WGS works in a simplified language”
Before 2000, it was impossible to read the entire genome of an organism in a day. It was a time-consuming, tedious and very costly process, at that time. However, thanks to NGS (next-generation sequencing), now it’s possible.
The massively parallel sequencing approaches available in modern times are capable enough to sequence the entire genome of any organism in under 24 hours. The NGS technologies therefore are widely used in clinical diagnosis setup. Unfortunately, whole-genome sequencing is still a challenging task!
I worked on NGS and sequencing platforms, did some substantial research and wrote dozens of good articles. From my personal experiences, I can say that it’s still difficult for students to understand how whole genome sequencing works, on a technical side.
In the previous article, we explained what genome sequencing is. In this article, I will explain the concept of whole genome sequencing, simply and understandably. This article is purposefully written for students and newbie researchers to make them understand the concept.
Stay tuned.
Disclaimer: The content presented herein has been compiled from reputable, peer-reviewed sources and is presented in an easy to understand manner for better comprehension. A comprehensive list of sources is provided after the article for reference.
Key Topics:
How does whole genome sequencing work?
As aforementioned, whole genome sequencing is a tedious, complex, lengthy, costly and time-consuming process. For instance, if we are talking about the human genome, consisting of 3.2 billion base pairs, it’s difficult to sequence such a long DNA fragment.
Previous generation sequencing platforms, such as Sanger sequencing take a few weeks to months to sequence the entire 3.2 billion bases with a decent sequencing depth.
The official time for the completion of the Human Genome Project (HGP) was around 10 years including data analysis. Now, imagine how hard and time-consuming whole genome sequencing is.
The reason why WGS is a difficult process is the presence of repetitive, homopolymeric and GC-rich regions. Because of this, it can not be sequenced, in a single stretch or run.
Myth buster!
If you think that whole genome sequencing is a continuous process, that means all 3.2 billion bases are sequenced in a single run! you are wrong. Understand why!
Any sequencer machine has sequencing limitations. It can effectively sequence a few hundred to a thousand bases. By the way, a machine’s sequencing capacity is known as “reads length.” To learn more read this article: What is ‘Sequencing Read’ in NGS?
For instance, the read length of Sanger sequencing is ~1000 bp, while the read length of Illumina sequencing is between 200 to 350 bp. Check out this table to learn more about the read length of various platforms.
| Platform | Sequencing generation | Read length (bp) |
| Sanger sequencing | First generation sequencing | ~1000 |
| Illumina | Second generation sequencing | 150-300 |
| Thermo Fisher (Ion Torrent) | Second generation sequencing | 200 to 400 |
| PacBio | Third generation sequencing | >50Kb |
| ONT (Oxford Nanopore Technology) | Third generation sequencing | >100Kb |
So to perform the WGS we need to chop down our genome into either 150 to 400-sized fragments (short-read sequencing) or 50 to 100 Kb-sized fragments (long-read sequencing). However, short-read sequencing is accurate and faster. To do so, we need to fragment our genome.
DNA fragmentation:
The preliminary step we have to perform just before the DNA extraction process is genome fragmentation. Noteworthy, depending on the platform’s read requirements, the NGS company provides the kit for DNA fragmentation.
But most certainly, it consists of a well-established restriction digestion enzymes combination.
After digestion, we have millions of random-sized DNA fragments. So you may have a question, how does the machine know which fragment to sequence? Because all the fragments, once sequenced, are also aligned back to generate the genome sequence. That’s where the known sequence adaptors help!
Adaptor ligation:
What we do is we ligate short and known DNA oligonucleotides to each fragment. On the solid sequencing surface, on which the sequencing will occur (flow cell or picotiter plate) the known oligonucleotide complementary sequences are immobilized.
So that the oligo-ligated fragment can settle there and provide a substrate for sequencing. These oligonucleotide sequences are known as adaptors. Although the adaptor length varies from company to company, the ideal length is >100 bp.
The adaptors also contain a few nucleotides long index or barcode sequences that are used for multiplexing different samples in a single run for high throughput sequencing. The machine or we can say the software knows all the adaptors and their index sequences and uses the data during the alignment process.
If you hear about the word “NGS library,” this is known as NGS or DNA libraries. It comprises all different-sized fragments ready for sequencing.
Sounds good! Let’s move ahead.
It might be possible that not all the fragments are uniformly fragmented during the library preparation process. And that will reduce the sequencing depth and thereby the overall accuracy.
What scientists do is, amplify the library. Using the random fragment amplification kit, the whole genome NGS libraries are amplified in the PCR machine. This will generate millions of copies for each particular fragment. In the next step, we will select fragments for sequencing.
This process is known as library enrichment in technical language.
Sequencing:
Now, here comes the main part- the sequencing process. The process usually occurs on a solid surface. The surface contains microscopic places, locations or wells (whatever we can call it!) on which adaptor complementary sequences are immobilized. Here is the list of various solid surfaces used by different companies.
| NGS Platform | Solid Surface | Unique Property |
| Illumina | Flow Cell | Single-molecule real-time (SMRT) sequencing, allowing for long read lengths and direct detection of DNA synthesis |
| Ion Torrent | Ion semiconductor chip | Detection of pH changes as nucleotides are incorporated, enabling real-time sequencing |
| Pacific Biosciences (PacBio) | Zero-Mode Waveguides (ZMWs) | Single molecule real-time (SMRT) sequencing, allowing for long read lengths and direct detection of DNA synthesis |
| Oxford Nanopore | Nanopore membrane | Detection of changes in electrical current as DNA passes through nanopores, enabling real-time sequencing |
| BGISEQ (Complete Genomics) | Combinatorial Probe-Anchor Synthesis (cPAS) arrays | Utilizes combinatorial probe-anchor synthesis for high-throughput and low-cost sequencing |
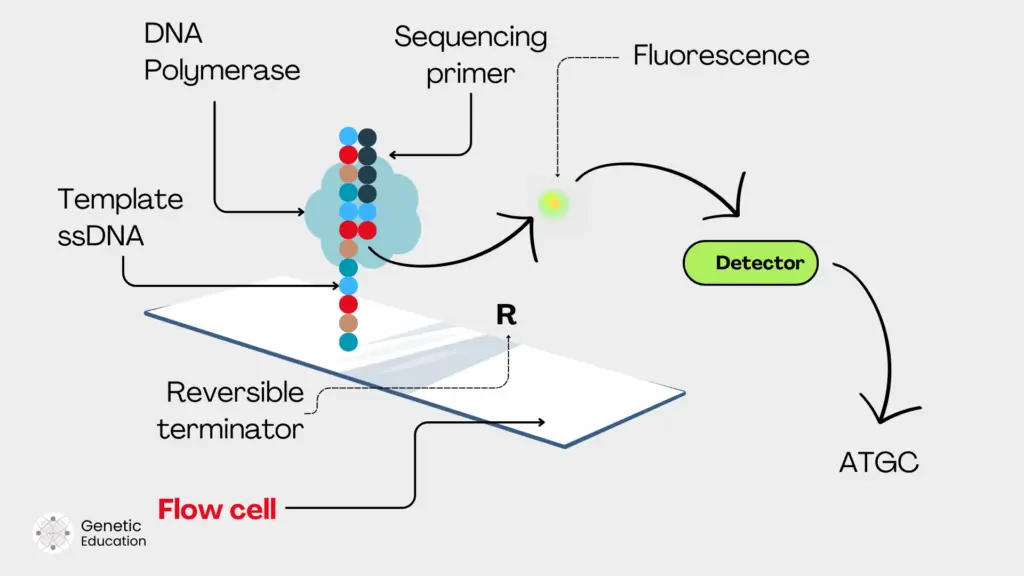
The common chemistry for NGS platforms is sequencing by synthesis. Meaning, that the sequencing process occurs during the DNA synthesis. When polymerase adds a nucleotide, it provides a signal, the detector detects the signal and converts it into a readable sequence.
Signals may vary from platform to platform including fluorescent signals or electrical signals. Various platforms and their signaling system are enlisted here.
| NGS platform | Detection chemistry |
| Illumina | Fluorescent |
| Ion Torrent | Ion semiconductor (electrical current) |
| PacBio | Fluorescent |
| ONT | Fluorescent |
For instance, when a fluorescently labeled nucleotide is incorporated by DNA polymerase, the fluorochrome is detached, the fluorescence is released, captured by the camera, detected by the detector and recorded as a positive signal. Usually, four different fluorescent tags are used for four different nucleotides.
Now, all the fragments are sequenced in a massively parallel process and a raw read file is generated. That file is known as the FASTQ file and is processed for the alignment process with the reference genome.
Reads alignment:
Based on the adaptor locations and read overlapping the computer program or software aligns each fragment in its correct location. To improve the accuracy, each read is aligned with the reference genome.
During the reads alignment process, first bad quality reads are trimmed off or removed by the software. And use only high-quality and correctly sequenced reads for the alignment. That’s entirely a software-based analysis.
Data analysis:
After completion of the alignment process, we have the complete sequence of our whole genome sequence. The data we can use for various studies and analyses. We can identify genome-wide SNPs, indels, disease-causing genes, etc.
Wrapping up:
This is the actual technical process of how whole genome sequencing is performed. The major steps in the present process are DNA extraction, library preparation, sequencing, alignment and analysis.
I guess, I have explained it effectively. If you want to strengthen your knowledge in the field of next-generation sequencing. You can join our online course at our Genetic Education Academy. The basic course price is 3200 but available at 999/- for early bird registration.The classes will start soon.
>> You can register from here: Next Generation Mastery Course.
I hope you like this article. To strengthen your NGS knowledge you can read other related. The links are already provided in the article. Do subscribe to our blog and share this article.
Sources:
Ng PC, Kirkness EF. Whole genome sequencing. Methods Mol Biol. 2010;628:215-26. doi: 10.1007/978-1-60327-367-1_12.