“A sequencing read is referred to as nucleotides of a single DNA fragment from a library. Sequencing reads, read lengths, numbers, read alignments and read scores are important terminologies, technically. Let’s see what these terms are.”
DNA sequencing, especially, the NGS and relevant technologies are yet to emerge fully but provide us valuable data. These next-generation sequencing techniques are by far more advanced, robust and speedy than the first or second-generation techniques.
Recent advances are so valuable that they can not only sequence the DNA or gene but also enable scientists to sequence the whole genome. Although to sequence, the entire eukaryotic genome needs extensive prerequisites.
Some such prerequisites are tagmentation including library preparation, adaptor ligations and barcoding. If you have some hands-on experience, you might know about these terms, if not you can read this segment from our previous article: Next-generation sequencing.
Anyway, the first-generation sequencing was a bit straightforward but has limitations, it can’t read longer sequences effectively, takes more time and is quite inaccurate. Wherein for NGS, above-listed prerequisites boost the experiment, read time, and read length, and thereby increasing the speed, accuracy and performance.
The fragmentation generates ‘reads’ for sequencing. ‘Reads’ is so crucial for the genomics and sequencing field. Some common questions one may have in mind are what exactly the sequencing reads are and what is the meaning of related terms.
Frequently, such terminologies create chaos and confusion for students and even for researchers. When you thoroughly understand it, it will help you to gain excellence in expertise in such technologies.
In this article, I will explain sequencing reads, read-time, and read-length and what are the implications of short and long reads on overall results. I will also explain how sequencing reads can be improved.
Moreover, understanding of NGS language would perhaps help scientists to communicate with clinicians, other non-technical persons, clients, patients and other scientists and make them understand what happens in NGS.
Using all of my previous experiences, and expertise in the field of sequencing, I will try to answer these questions and many more relevant ones. Stay tuned.
Related articles:
- DNA Sequencing: History, Steps, Methods, Applications and Limitations.
- What is Genome Sequencing?- 3 Best Genome Sequencing Methods
Key Topics:
What is sequencing read?
‘Reads’ is the terminology of NGS costume language.
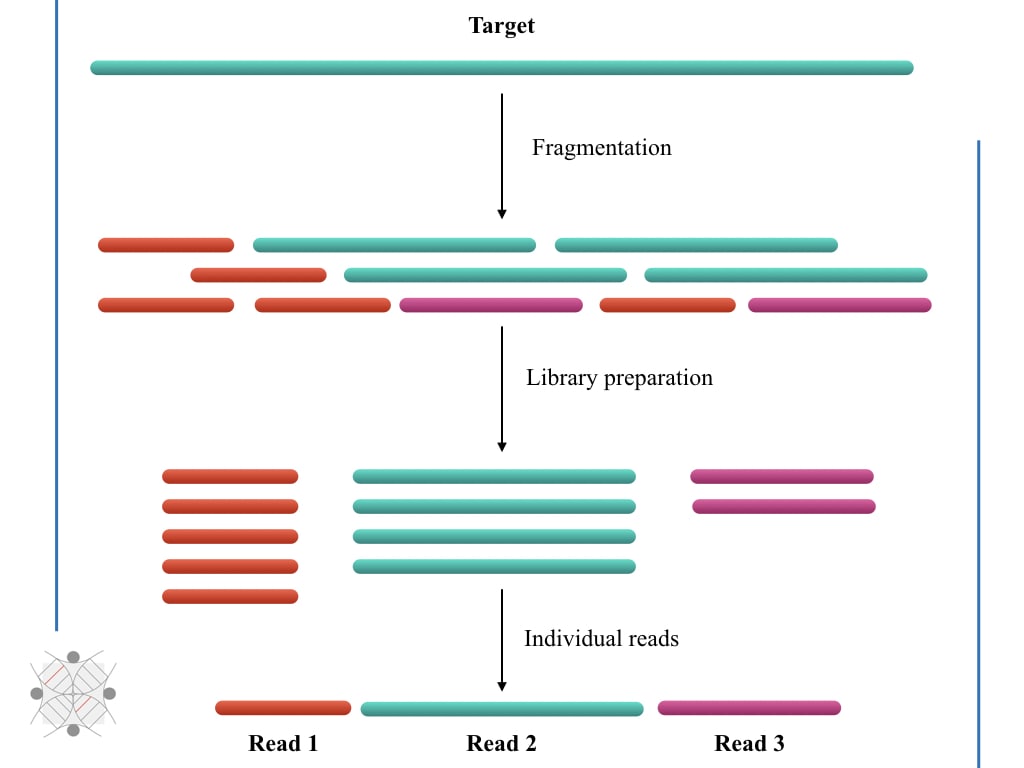
To sequence huge genomic portions or the entire genome, it should be cleaved into fragments. Because the entire genome can’t be effectively sequenced in a single run, it’s huge. Techniques like restriction digestion do the job.
Incubating high-quality genomic DNA with a specific or provided restriction enzyme will generate random and map-able sized genomic fragments. Fragments range from 100 to 100kb, depending on the need for the experiment.
Such fragments, to make it recognizable are ligated with known sequence adaptors which eventually would help to re-sequence it back. It is known as a genomic or fragment library.
Sequencing reads in NGS is referred to as, “sequencing of all nucleotides of a fragment from a library.”
Let us comprehensively interpret it. A genomic library, as we aforementioned, is a group of known, recognizable and same-sized fragments. Notedly, as restriction enzymes have randomly located restriction sites in a genome, they produce random-sized fragments.
For example, 100bp fragments are library 1, 150bp fragments are library 2 and 900bp fragments are library 3, etc. It can generate numerous same-sized fragments, and the numbers even can be increased by PCR amplification.
The sole purpose of increasing the number of fragments or generating copies of a specific fragment library is to understand and increase the quality of read depth. Now, I think you understand that when the machine reads a single 100bp fragment from the library of 100bp fragments, it counts as “read 1”. And so on.
Definition of sequencing reads:
Sequencing read is referred to as, “the sequencing of nucleotides from a single fragment of a library.”
Read length:
The read length is the entire length or size of a particular read. Read length is a significantly crucial factor of any NGS and varies depending upon the applications, sample and requirements of the client.
In this context, two common read types are long sequencing reads and short sequencing reads. Short reads are typically between 80 to 200 bp whilst long reads are 500 to 2.3Mb in size. Importantly, long reads have more resolution power than short reads and are often practiced for whole genome sequencing.
| Technology | Read length |
| Sanger sequencing | 500 to 600 bp (Max up to 900bp) |
| Maxam Gilbert sequencing | 500 to 600bp |
| Sequencing by synthesis | 100 paired end |
| Pyrosequencing | 1000bp |
| ABI SOLiD (Colour space) | 60 paired end |
| HiSeq4000 (Illumina) | 150 bp paired end |
| iSeq 100 (Illumina) | 150 bp paired end |
| NovaSeq (Illumina) | 150 bp paired end |
| MiSeq (Illumina) | 300 bp paired end |
Sequencing read time:
Yet another important terminology in the NGS world is the read time. It is important, how much time the machine sequences a read, it matters. A typical NGS experiment takes 24 hours for up to 700Mb sequence analysis in a day.
Sequencing short vs long reads:
| Short sequencing reads | Long sequencing reads |
| Read size is 80 to 200bp | Read size is as low as 500bp and as long as 2.3Mb. |
| Read time is less. | Read time is more. |
| Sequencing by synthesis and sequencing by ligation. | Depending on the instrument |
| Generates fewer overlaps. | Generates overlaps. |
| Low sequencing capacity for highly repetitive telomeric or centromeric regions, and the entire genome. | High sequencing capacity for highly repetitive telomeric or centromeric regions, and the entire genome. |
| Some examples are Illumina, 454 pyroseq, Ion torrent, SOLiD and cPAL. | Some examples are Single molecular real-time sequencer technology (SMRT) and nanopore sequencing and conventional Sanger sequencing. |
| Low error rate and highly accurate. | High error rate, less accurate. |
| Can’t find genome-wide rearrangements. | Improve the resolution of de novo assembly and finds genomic rearrangements. |
Important notes:
Advances in technology show that long reads can efficiently read 5,000 to 3,000 nucleotides in a single run. For example, the SMRT sequencer can read approximately 10Kb fragments in just two hours. Ulta short reads: reads of 20 to 40 bp are denoted as ultra-short reads and are not useful for repetitive region sequencing. All NGS platforms produce short reads of up to 200bp while Sanger sequencing produces longer reads. (Add a read-length graph here using the above data).
Read lengths of different generation sequencing:
| First generation sequencing | 200 to 600 bp long reads |
| Next generation sequencing | 80 to 200bp short reads |
| Third generation sequencing | 5kb to 2.3 Mb ultra-long reads |
Sequencing read overlap:
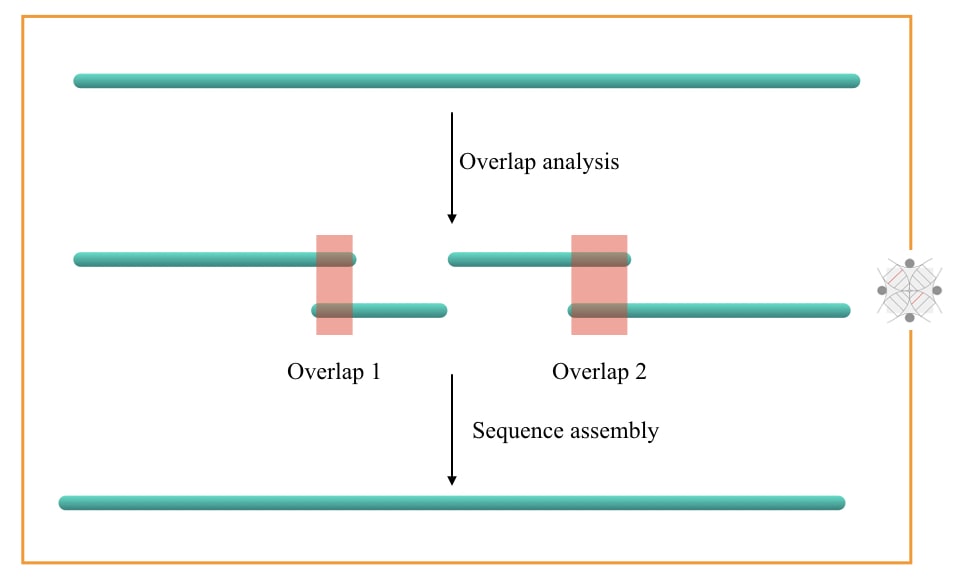
Sequencing read overlap is a problem that occurs during long-read and even short-read NGS sequencing. It is when a portion or nucleotide sequence of one read overlaps with another larger read. It has important implications before the final read or genome assembly.
A separate computational tool analysis reads and highlights possible overlap regions if exist. Such overlap between two reads can be merged using the reference sequence data. Read overlap analysis will increase the efficiency of reading assembly.
The algorithms used to determine the read overlap are MHAP- MinHash Algorithm and BLASR. Besides, another algorithm often used for long read overlap is LROD (Long read overlap detection). Both are based on the K-mer distribution.
Sequencing read assembly:
Put simply, sequence read assembly is a process of assembling or arranging all the reads in the correct order. It is often known as genome assembly, read assembly or sequence assembly.
Technically, it’s a software-based computational method to merge correct reads with each other, thereby constructing the original nucleotide order of a genome or target that we wish to study. However, it is important to know that millions of individual reads are generated, aligned and assembled to generate the entire genome sequence.
Sequencing read assembly is an important and interesting topic and should be discussed separately. I will write a dedicated article on it and update it here.
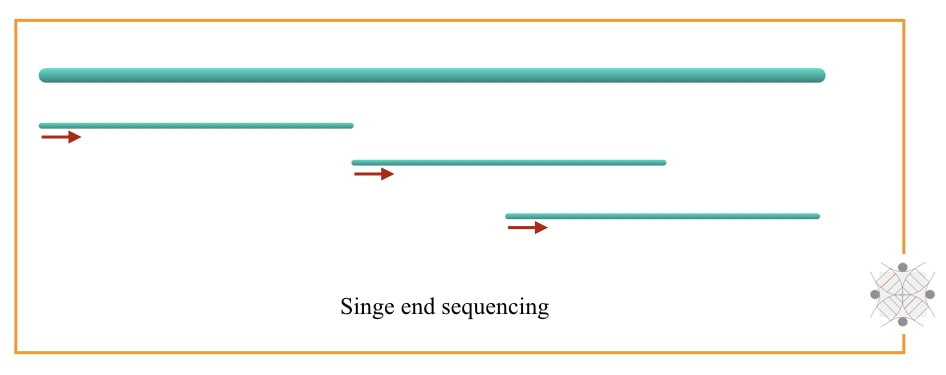
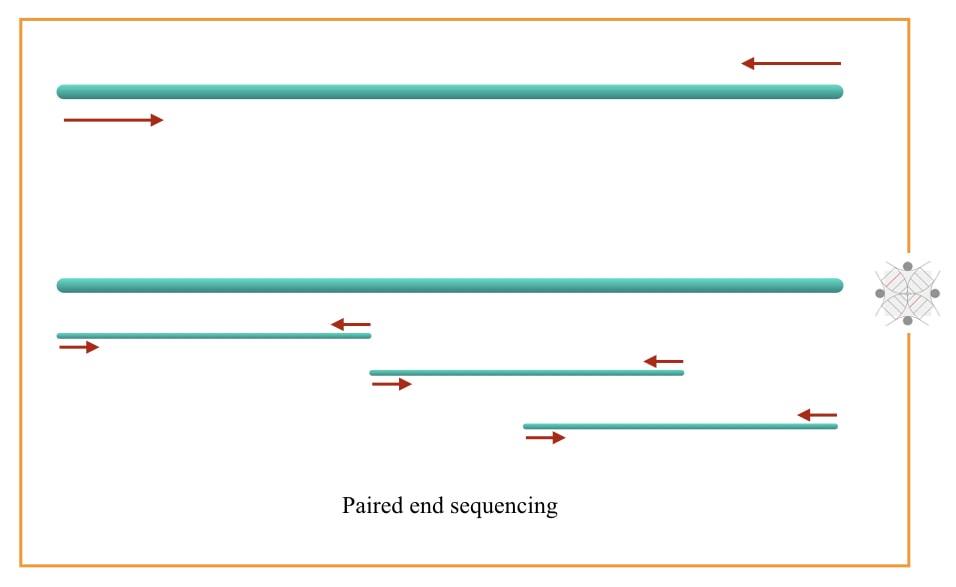
Paired vs single-end sequencing reads:
Paired-end sequencing sequences the read (fragment) from both ends while single-end sequencing reads sequence the read from a single side or one end. So it is understandable that paired-end sequencing provides more accurate results.
The reason is clear, it sequences the fragment and re-sequences it again. But unfortunately, it is costlier.
Paired-end sequencing is not for all types of sequences. It is often employed for special sequence characters like to read repetitive sequences, genomic rearrangements and gene fusion etc. Such aberrations are so complicated in nature and require expertise to interpret.
On a technical note, it can accurately find insertions and deletions thereby novel copy number variations in the genome. So it is often and only employed for genomic rearrangements. studies. On the downside, paired-end sequencing is a costly, time-consuming and tedious process.
Single-end sequencing reads the sequence of the fragment in a unidirectional. For example, the conventional Sanger sequencing approach. Such an approach has poor resolution, is comparatively inaccurate and inadequate to study genomic rearrangements.
However, short sequences or gene alterations can effectively be studied using single-end reads. The present approach is simple, cost-effective and time-saving but at the same time has less accuracy.
Related article: Single-End vs Paired-End Sequencing.
Read depth:
Read depth is an important consideration in the NGS and is a crucial parameter having a direct effect on the overall sequencing results. It shows how many times a particular nucleotide or read is sequenced.
You might hear about 60X, 85X or 25X depth coverage of sequencing. It depicts that 60, 85 or 25 times a target nucleotide or read is sequenced. However, it is different from read to read and nucleotides to nucleotides.
For example, some are sequenced for 5 times (5X), and some are for 95 times (95X). So for 85 to 90% of nucleotides of our target should have read depth coverage of more than 15 to 20X.
Read mapping:
Read mapping is a process to map or align the nucleotide sequence generated from sequencing to the reference sequence. We can say, it’s a final step in the computational analysis and give us an idea about the correctness and present or absence of novel alterations within the sequence, target or genome.
There are software available for read mapping which is important to discuss. So we will cover an entire article separately.
How to improve sequencing read quality?
How many sequences, how many times and how accurately a machine reads, is entirely depends on the machine. We can’t do anything about this. But what we can do is, we can improve the performance of sequencing and by that the quality of reads.
- Extract a good quality DNA.
- Prepare a genome library well.
- Do excellent fragmentation.
- Screen libraries before NGS to check their quality.
- Performs amplification and enriches libraries well.
- Use high-quality reagents for sequencing.
- Prepare an excellent sequencing reaction or as instructed by the kit provider.
If you improve the overall read quality, the quality of the output obtained from every step, like the read length, read depth, assembly and mapping would eventually increase.
Wrapping up:
Next-generation sequencing is a complex, yet fast, accurate and sensitive sequencing platform. It can even sequence the entire genome of any eukaryotic organism within a week. And thus get penetration into genetic science.
However, NGS is costly, and a common person or independent research scholar can’t afford it. At the same time, some platforms have a higher error rate as well.
I hope the present article can help you in your sequencing learning journey. Share the article and bookmark the page.