“DNA sequencing is a process of determining or identifying the order of nucleotides present in a DNA sequence.”
DNA is a nucleic acid and genetic material of us. It is made up of nitrogenous bases, sugar and phosphate. Four nitrogenous bases in DNA are Adenine, Thymine, Cytosine and Guanine. Phosphodiester bonds join adjacent nucleotides while hydrogen bonds join nucleotides of opposite strands.
The whole DNA of a cell is known as a genome having two critical functions to encode proteins and regulate gene expression. These two goals are achieved by non-coding sequences and coding- genes, respectively.
Alteration in nucleotides forms a faulty gene thereby manufacturing an incorrect protein. In addition, polymorphisms, oftentimes, alter gene expression too. DNA sequencing allows us to understand the order of nucleotides, gene structure, alterations or mutations within, and so many other things.
Inherited genetic disorders, mutations, complex traits and other genomic phenomena can also be understood by DNA sequencing. So in brief, it helps us to perceive the knowledge of genome complexity. But how is it done? What are the steps and a few common methods?
Let us find out.
This article is huge, and comprehensive and contains all theoretical and practical knowledge on sequencing. I will discuss a short history, process and common methods of sequencing. Furthermore, this article will let you understand how you can sequence DNA and read it.
This lesson would be helpful in your DNA sequencing endeavors. So take a cup of coffee, a pen and paper, and stay tuned.
Key Topics:
What is DNA sequencing?
“Put simply, it is a process of determining the sequence of nucleotides.” Now first understand why sequencing is required.
PCR has a serious limitation, as it can only determine known sequence variations which are indeed not sufficient and hard to identify, sometimes. It can’t even read nucleotides.
Further to this, it lacks the potential to denote novel sequence variance or polymorphism without which faulty gene or altered expression can’t be studied.
Thus we need a powerful technique that can read every nucleotide present in a DNA and let us know the order and alteration(s) if any. DNA, gene or genome sequencing can solve all of these problems.
Sequencing is a combination of both wet and dry lab work. A machine performs a chemical synthesis while a computer program performs sequence analysis. There are many sequencing platforms available, each one uses different chemistry and relies on a different principle for sequencing.
Definitions:
“A laboratory technique employed to determine the sequence of a gene or DNA by a sequential chemical reaction is referred to as DNA sequencing.”
Principle of DNA sequencing:
In general, a broad principle of DNA sequencing is as stated.
“DNA is denatured and converted into single-stranded. The sequence read by the sequencer machine gives signals to the detector which transfers them to the computer. A computer software inspects the signals and converts each read into a nucleotide sequence.”
Note that this is an in-general principle to understand the sequencing process. Each technique has its own working principle that we will discuss in each separate segment.
History of DNA sequencing:
The story of DNA began when Watson and Crick discovered the structure of DNA in the year 1953. In 1964, Richard Holley and co-workers who performed the sequencing of the tRNA was the first attempt to sequence the nucleic acid.
They sequenced the RNA genome of the bacteriophage MS2 using the technique of Holley and Fieser W. So, the first nucleic acid sequenced was the RNA, not DNA. DNA was yet not sequenced. In the year 1977, Fredrick Sanger postulated and published the first method for sequencing DNA- a chain termination method.
In the following year, Maxam A and Gilbert W postulated the chemical degradation method of DNA sequencing. The genome of bacteriophage X174 was sequenced in the same year using the chemical degradation method proposed by Maxam and Gilbert.
Both techniques (chain termination and chemical degradation) were laborious, tedious, time-consuming and lacked automation. The first semi-automated DNA sequencing method was developed by Lorey and Smith in 1986.
Later on, in 1987, Applied Biosystem developed a fully automated machine-controlled DNA sequencing method. After the development of fully automated machines, the era of the 2000s became a golden period for sequencing platforms.
Furthermore, in 1996, Applied Biosystem developed another innovative sequencing platform known as capillary DNA sequencing which was based on the principle of capillary electrophoresis. Interestingly, these two automation in sequencing has a pivotal role in the completion of the human genome project which was later completed in 2003.
A fast, accurate, reliable, and highly efficient next-generation sequencing platform was postulated in the year 2005 by Solexa/Illumina. Some of the milestones in DNA sequencing are shown in the figure below,
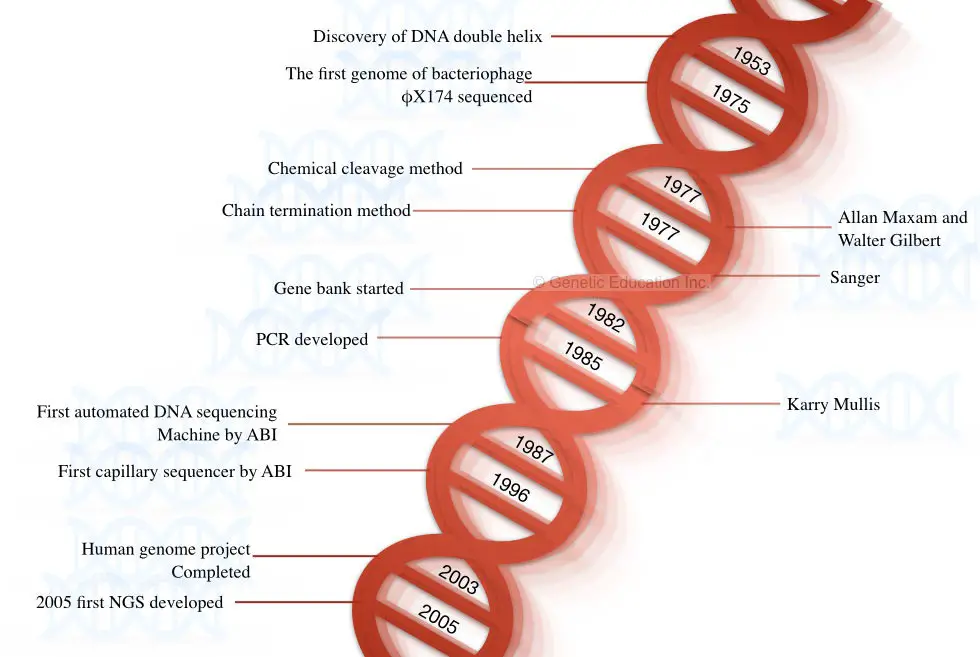
Steps and Process in DNA sequencing:
This segment answers the question: what are the different steps in DNA sequencing? Note that the steps enlisted and explained below are a generalized scheme. The principle, steps and process vary from sequencing platform to platform.
- Sample preparation- DNA extraction
- DNA fragmentation
- Adapter ligation and Library Preparation
- Library enrichment- amplification
- Library purification
- DNA sequencing
- Data collection and sequence analysis
Sample preparation:
Sample preparation starts with extracting pure and high-yield DNA. DNA of any organism, for example, animal, human, plant, bacteria or virus can be sequenced in the same process which begins with DNA extraction.
However, DNA extraction techniques vary between different organisms. To learn more you can follow this article: 10 different types of DNA extraction methods.
Ideally, the concentration and purity of DNA for sequencing are ~50 to 500 ng and 1.80 (260/280 ratio), respectively. One should follow the instructions and suggestions provided by the manufacturers.
DNA fragmentation:
For a larger sequence or gene or whole genome, the DNA fragmentation step is so important. Here, using enzymatic catalytic reaction, chemical or physical treatment, the large chunks of DNA, which can not be sequenced, are fragmented.
Small fragments enable accurate reading during sequencing. Generally, restriction digestion is performed to prepare smaller DNA fragments of 1000 to 10,000bp. Fragments are saved in a library.
Adapter ligation and library preparation:
Sticky ends generated by the fragmentation are ligated with the known adaptor sequence and sometimes with a known barcode sequence too. After completion of ligation, the same-sized fragments are stored in the genomic library.
Adaptors and known oligonucleotide barcodes help in amplification and re-arrangement of sequence after accomplishment of the process.
Note: If the desired fragment is too short of up to 100bp, fragmentation and library preparation are not required, for example, 16s rRNA or 18s rRNA gene sequencing.
Do you know that the cDNA library also exists? To know more about it, refer to this article: cDNA library preparation.
Library enrichment- amplification:
To sequence the desired fragment, we need millions of copies of that particular fragment or a whole library. PCR amplification generates copies and facilitates library enrichment.
The amplification completes in three steps denaturation at 94°C, annealing at 55°C to 65°C and extension at 72°C for 35 cycles. The enriched library is purified, later on.
Library purification:
Why is purification needed?
This information is very interesting, no one will tell you about it.
If you check the purity of the PCR product at 260 and 280nm wavelengths, it will always be 1.80, exactly! because the amplicons are pure DNA fragments. If any contaminants are present in the DNA, amplification will not occur. Then why is amplicon purification required? the answer is here,
Unbound primers, primer-dimers, unused Taq DNA polymerase, unused DNA templates, and other unused PCR reaction buffer components can abort DNA sequencing. That is why amplicon purification is required.
Here, alcohol purification can’t work. To achieve maximum purification and amplification yield, we need to use a ready-to-use amplicon purification kit. Such kits use spin-column technology which is handy, fast and accurate. The purified library is ready for sequencing.
DNA sequencing:
Keep in mind that the sequencing platform and chemistry vary from machine to machine. Some common chemistry is chain termination, chemical degradation, bridge amplification, etc.
Put simply, in a sequencing machine, the machine reads each nucleotide of a target sequence, the detector detects signals and sends them to the computer. For example, let’s take the example of sanger chain termination.
When a ddNTP is incorporated during amplification, the synthesis of the nucleotide chain is terminated, fluorescence is released, the detector detects the signal and sends it to the computer.
Data collection and sequence analysis:
In the final step, the sequencing file is processed in a computer program. The inbuilt software (provided by the manufacturer) processes the data and compares it with the available sequence information.
Through pairwise alignment, each nucleotide is compared with the reference, and any variations within the sequence are noted, reported and documented. The entire process of DNA sequencing is explained in the figure below.
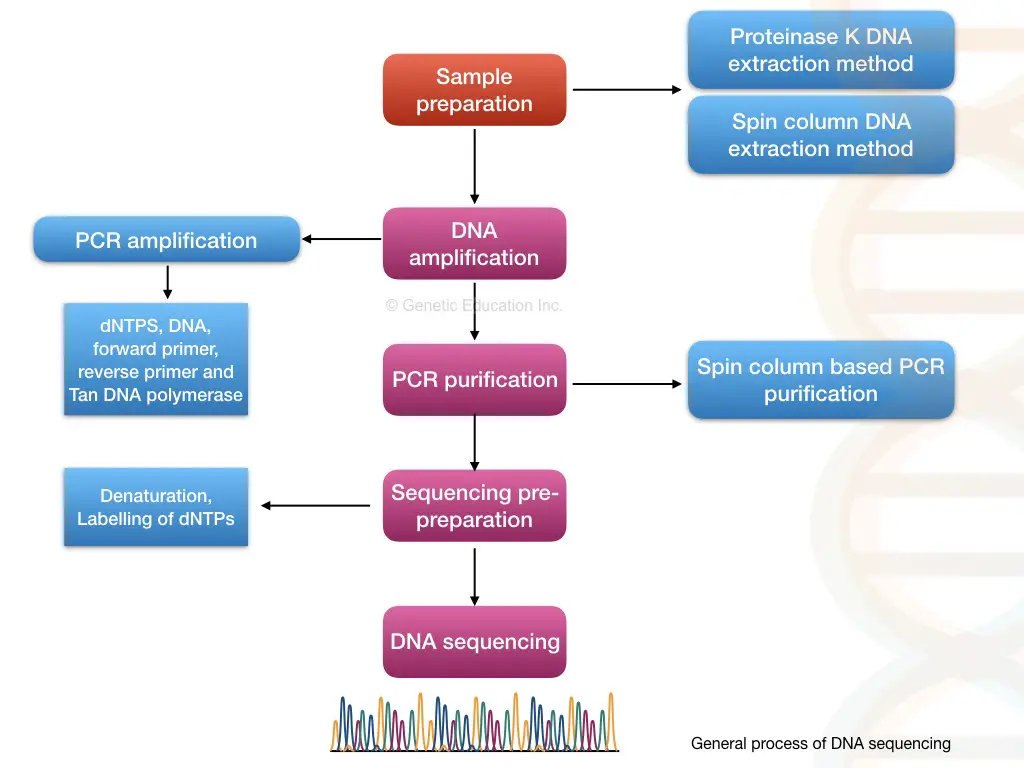
Different methods of DNA sequencing:
In this section of the article, we will discuss some of the common and important methods of DNA sequencing. Take a look at the list first.
- Maxam and Gilbert’s method
- Chain termination (Sanger sequencing) method
- Semiautomated method
- Automated method
- Pyrosequencing
- Whole-genome shotgun sequencing method
- Clone by the clone sequencing method
- Next-generation sequencing method
Terminologies used in the article:
| Term | Explanation |
| Fragment library | A collection of the entire strand of the DNA (to be sequenced) fragments. |
| Gaps | Here the un-sequenced region of the DNA is called a gap. |
| Conting | A continuous sequence of the DNA is assembled. |
| Read | The output data came from the sequencer machine to the computer for one particular sequence. |
| Coverage | The number of times the sequencing machine covered the DNA sequence. |
Maxam-Gilbert sequencing:
- Year: 1977
- Scientists: Maxam and Gilbert
- Names: Maxam and Gilbert sequencing, chemical degradation sequencing.
- Automation: Not possible.
The first 24 nucleotides, in the history of genetics, were sequenced by Maxam and Gilbert in 1977. The method developed by these two is also known as the chemical cleavage method as well. Unfortunately, they are not the pioneer in sequencing, as their method was published after two years of the Sanger sequencing method.
The brief principle of the present method is as stated,
Principle:
Chemical degradation is performed on a single-stranded DNA using selective chemical treatment (hydrazine and hydrazine NaCl for pyrimidine and dimethyl sulfate and piperidine for purine). The fragments generated by the treatment are run on a polyacrylamide gel to separate and identify them.
Process:
It starts with DNA extraction. Pure DNA is heat denatured and converted into a single-stranded one. The process is followed by dephosphorylation and radiolabeling.
An enzyme named phosphatase removes the phosphate from the 5’ end while the kinase adds the 32P to the 5’ end of DNA, simultaneously.
4 different chemicals cleave DNA at four different positions; hydrazine and hydrazine NaCl selectively attack pyrimidine nucleotides while dimethyl sulfate and piperidine attack purine nucleotides. Take a look at the chemistry here.
- Hydrazine: T + C
- Hydrazine NaCl: C
- Dimethyl sulfate: A + G
- Piperidine: G
Denatured DNA of equal volume is added to 4 separate tubes containing these 4 different chemicals. Afterward, sample incubation is performed followed by gel electrophoresis using PAGE- polyacrylamide gel electrophoresis.
The results of chemical cleavage are shown in the illustration given below. To enable visualization autoradiography analysis is carried out. The presence of 32P on the DNA allows us to determine fragments generated by the cleavage.
Applications:
The Maxam and Gilbert technique is more accurate than Sanger sequencing. It’s best suitable for DNA footprinting and DNA structural studies. It is more advantageous than the Sanger method as it uses purified DNA directly for sequencing.
In the present automation time, the present method is used in DNA fingerprinting and genetic engineering studies.
Notwithstanding, plenty of disadvantages make it harder to use regularly. It comes up with poor scalability, only up to 400bp sequence can be read. Moreover, It is less popular because of the use of harmful radiolabeled chemicals.
| Advantages | Limitations |
| Base-specific cleavage.Does not require polymerase to synthesize DNA. Sequence purified DNA directly Allow protein DNA interaction studies. Easy to use, handy and cost-effective. | Limited sequencing capacity of up to 400bp only. Relies on radiolabeling which is harmful. Uses toxic chemicals in the process. Can’t be scaled up. |
Sanger sequencing:
- Year: 1977
- Scientists: Sanger
- Names: Sanger sequencing, chain termination sequencing, first-generation sequencing and dideoxynucleotide sequencing.
- Automation: Possible.
| Sequencing platform | Read length | High throughput | Accuracy | Run time |
| Sanger sequencing | 400 to 900bp | <3 Mb | 99.9% | 4 hours |
Sanger and co-workers developed a chain termination method of DNA sequencing, after a few years of Maxam and Gilbert’s method. It is also known as the first-generation DNA sequencing method. The present Technique is often referred to as dideoxynucleotide sequencing as it uses specially synthesized nucleotides- ddNTPs. Notedly, the dideoxynucleotides possess a hydrogen group instead of a hydroxyl group. It makes ddNTPs different from normal dNTPs.
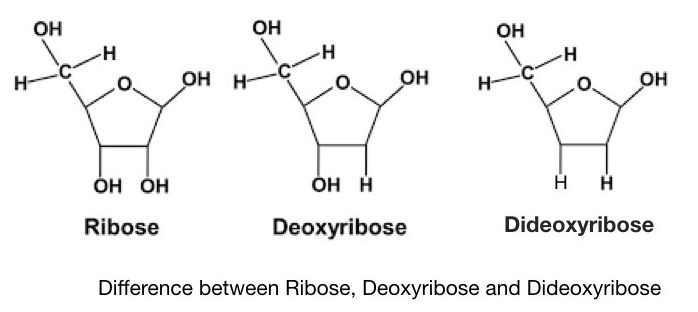
Principle:
A denatured DNA is synthesized by DNA polymerase by adding dNTPs but when it encounters the ddNTPs, the presence of hydrogen (instead of hydroxyl group) terminates the chain synthesis. Each ddNTPs terminate the chain when encountering a complementary nucleotide every time. The results are analyzed on a PAGE gel which distinguishes each termination by length differences.
From a technical perspective,
The Phosphodiester bond can’t form between two adjacent nucleotides due to the absence of a hydroxyl group in ddNTPs. This restricts the synthesis and henceforth, the technique is known as the chain termination method.
Do you know?
Sanger sequencing technique was utilized in the human genome project.
Process:
The process of Sanger sequencing is broadly divided into 5 steps:
- DNA extraction– using any of the DNA extraction protocols.
- DNA denaturation- using heat or chemical treatment.
- DNA synthesis– using the flanking primers, dNTPs, Taq DNA polymerase and PCR buffer.
- Cycle sequencing- using all dNTPs, ddNTPs, Taq DNA polymerase and PCR buffer.
- Separation of fragments by size- using PAGE, or capillary gel electrophoresis.
In the very first step, DNA isolation is achieved. DNA extraction is followed by DNA purification and DNA denaturation. Any DNA extraction technique can be used here, however, the goal is to isolate pure and good-quality DNA.
DNA sample with a purity of 1.8 and quantity of 100 ng is selected and processed further. In the next step, the PCR performs amplification to get copies of the desired fragment.
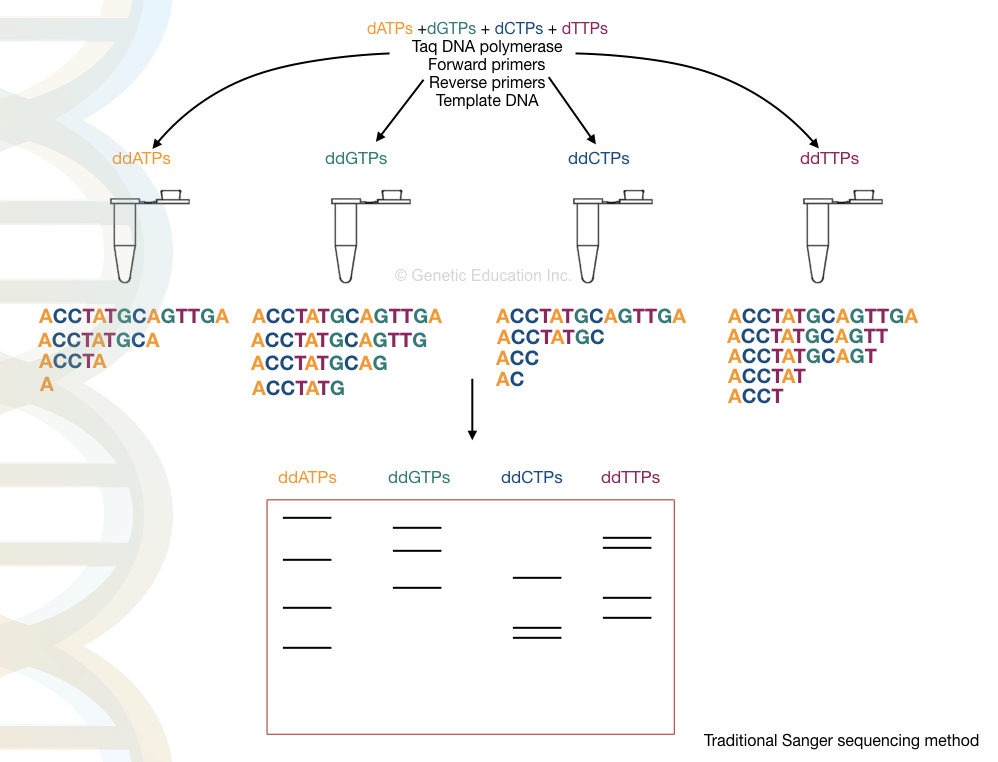
In the next step, the cycle sequencing is performed by using the denatured PCR products. The reaction preparation is shown here.
| Reaction | PCR reaction | modification |
| Reaction “A” | Taq DNA polymerase, dATPs, dGTPs, dCTPs, dGTPs and PCR buffer, primers | Labeled ddATPs |
| Reaction “G” | Taq DNA polymerase, dATPs, dGTPs, dCTPs, dGTPs and PCR buffer, primers | Labeled ddGTPs |
| Reaction “T” | Taq DNA polymerase, dATPs, dGTPs, dCTPs, dGTPs and PCR buffer, primers | Labeled ddTTPs |
| Reaction “C” | Taq DNA polymerase, dATPs, dGTPs, dCTPs, dGTPs and PCR buffer, primers | Labeled ddCTPs |
Each tube contains the same amount of PCR reagents but in each tube, extra ddNTPs are added as shown in the table. Here the flanking primers bind near the region of a sequence of our interest.
In the cyclic reaction, the Taq DNA polymerase incorporates nucleotides- dNTP on a denatured DNA. Here, high fidelity DNA polymerase is not needed. Normal Taq polymerase expands the growing DNA strand by the addition of the dNTPs.
Interestingly, once it adds the ddNTP instead of dNTP the chain expansion is stopped or terminated.
The termination process is complete in 4 different tubes for 4 different ddNTPs for each nucleotide. For example, in the ddATP tube, termination occurs at all positions where ddATPs get bound.
Final amplicons from each tube run on a polyacrylamide gel which separates DNA fragments based on their size. Note that smaller fragments run faster than larger ones.
The final results are analyzed in a UV transilluminator, gel doc or X-ray film. We have hypothetically prepared the reaction, run it and analyzed the results. Take a look at it.
Advancements in Sanger sequencing:
Sanger sequencing is the gold standard method for research as well as for diagnosis, nowadays because of its easy setup and high reproducibility. In addition, automation also makes it more usable in routine.
Traditionally, the results are interpreted on PAGE manually but now, the scenario is changed. The process is fully or partially automated. Instead of normal ddNTPs, fluoro-labeled ddNTPs are used.
When it binds, the fluorochrome releases fluorescence, and a detector detects signals each time when the chain is terminated. Further, the signals are recorded and analyzed computationally.
Samples are run on an automated electrophoresis device known as capillary electrophoresis having the power to distinguish a single nucleotide change very precisely. In the process, samples are run in long capillaries and fluorescence is detected when it leaves the capillary.
The computational software generates various fluorescence peaks depending upon the amount of fluorescence emitted. The hypothetical illustration of the Sanger sequencing results is shown in the figure below,
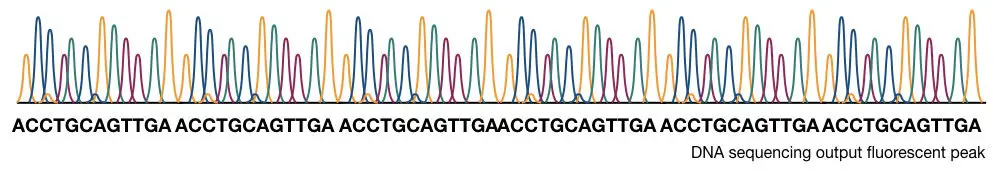
How to interpret the PAGE result for Sanger sequencing?
This section is very important, I will teach you how you can interpret the results of the sequencing. Although it is a type of overview.
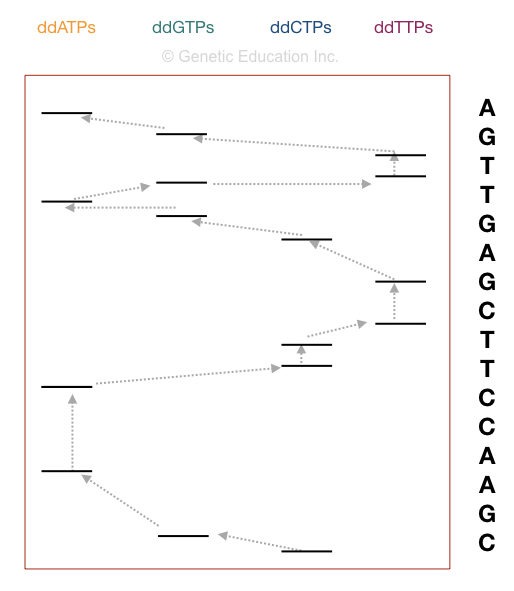
Now take a look at the figure above,
The principle of electrophoresis stated that the smaller DNA fragments migrate faster than larger ones. So the fragment on the positive side is the smallest one. Start reading the sequence from that.
The first point in sequencing the DNA is to match the size of DNA. if the bands obtained in gel and the nucleotide sequence of our DNA are similar, the reaction is completed properly. For instance, if your sequence length is 16, then 16 bands must be present in the gel.
Start reading from the bottom. The DNA sequence starts with “C” as the last fragment of DNA terminated with ddCTP. Arrange the sequencing shown in the figure, accordingly.
Automated Sanger sequencing:
The automated Sanger sequencing is a combination of amplification, capillary gel electrophoresis and detection by various techniques. The conventional Sanger technique is a tedious process, but recent advancement makes it easy and rapid to use.
The semi-automated Sanger sequencing method is based on the principle of Sanger’s method with some minor variations. Automation occurs in two ways, which also depend on the chemistry used by the manufacturer.
Fluorescent labeling of each ddNPTs with four different fluorochromes and run in a single capillary. And labeling of flanking primers with a single dye and running in four separate capillaries. However, for the second option, the detection technique used is an argon ion laser beam.
The samples are run in the automated CE setup and separate. By using the labeled primers or the ddNTPs, the machine reads the sequence accurately, on single-lane capillary electrophoresis. We can sequence more than 300 samples in a single run on an automated DNA sequencing platform.
The capillary electrophoresis is used to separate DNA molecules on the basis of size, It is powerful enough to separate a single base pair fragment. The chromatogram generated through the C.E sent the output as a fluorescent peak. What results it gives are known as electropherograms. If you want to learn what an electropherogram is and how to read it. Please click the link and refer to the article. Roughly, the process of it is explained below here,
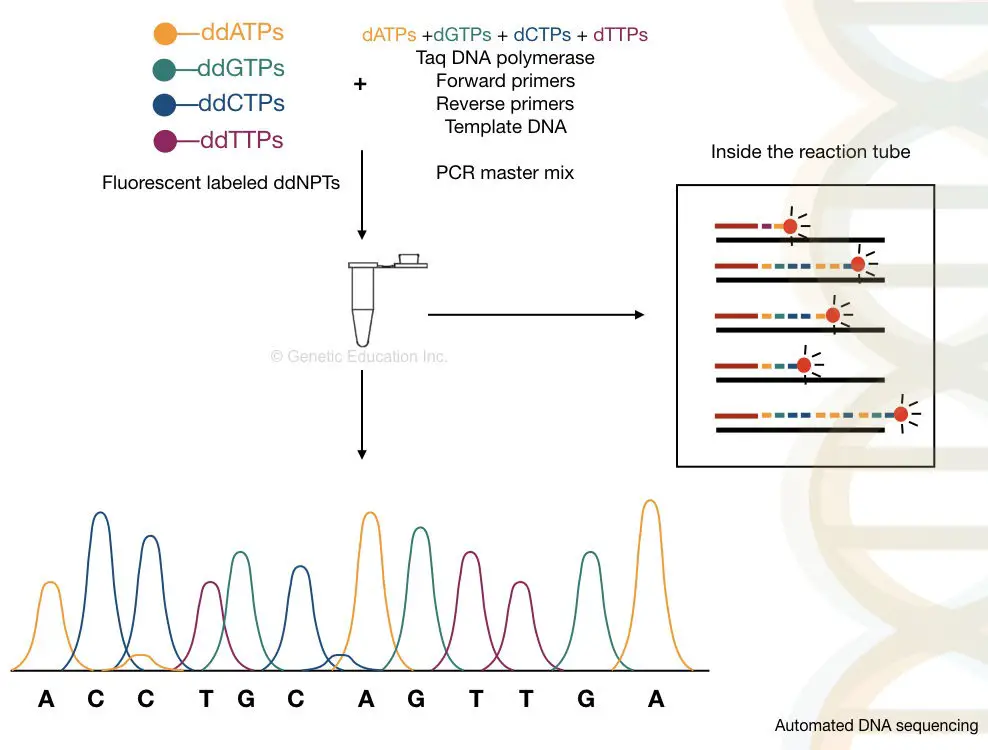
Take a close look in a tube.
What happened inside the tube?
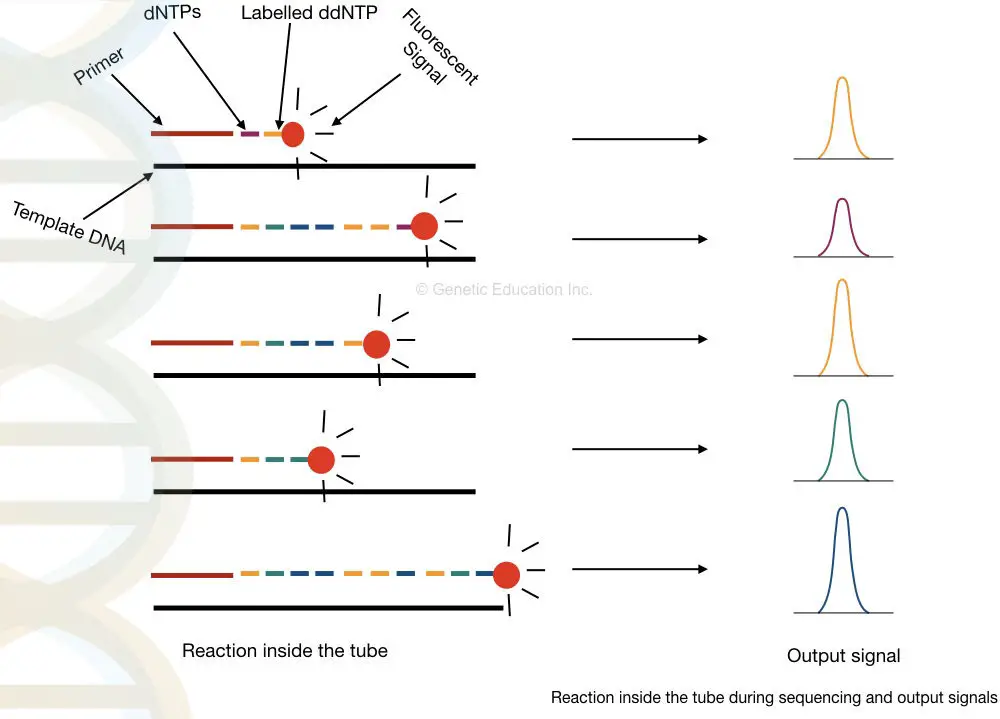
Applications
Sanger and automated Sanger sequencing is the most anticipated, widely used and trusted technique to sequence DNA. Broadly, some of the important applications are enlisted here.
- Sequence both, prokaryotic and eukaryotic genomes.
- Sequence a gene.
- Microbial identification by 16s rRNA gene sequencing.
- Mutations and mutagenesis studies.
- Clinical and research studies.
- In recent times, robust and automated sanger sequencing helped to map the genome of SARS-CoV-2 COVID19.
- Used in identification, characterization and classification of microbes.
| Advantages | Limitations |
| More accurate, robust and automated.State-of-art and high throughput. More read capacity than the chemical degradation method. Read 700 to 800 bp in a single run, accurately. Low error rate. Low turnaround time. | Time-consuming and costly. Not suitable for long gene sequence, genes and genome sequencing. Required advanced and costly infrastructure. |
Sequencing fact
It takes approximately 50 years to sequence all the 3.2 billion bases of the human genome through the semi-automated Sanger sequencing method.
Pyrosequencing:
- Year: 1993
- Scientists: Bertil Pettersson, Mathias Uhlen and Pål Nyren
- Other Names: –
- Automation: Possible.
| Sequencing platform | Read length | High throughput | Accuracy | Run time |
| Pyrosequencing | ~500-700bp | 700mb per day | 99.9% | 8 to 23 hours |
In 1993, Bertil Pettersson, Mathias Uhlen and Pål Nyren described the pyrosequencing method. It is a type of solid phase sequencing that uses magnetic beads coated with streptavidin.
However, solid-liquid phase pyrosequencing is also available.
Principle:
The present technique relies on the principle of sequencing by synthesis using DNA polymerase which lacks proofreading.
The method is based on the detection of the pyrophosphate released during the chain reaction of nucleotide addition. Here the order of the nucleotide is determined by the PPi released during the joining of two adjacent nucleotides (3’OH- 5’P).
Process:
In contrast with other methods, along with a polymerase, two additional enzymes are required in the pyrosequencing method. The three enzymes are
- DNA polymerase (without exonuclease activity)
- Luciferase
- Sulfurylase
All three enzymes work in a sequential manner for the detection of the PPi. The real-time polymerase activity monitoring allows the detection of the released pyrophosphate in a cascade of the enzymatic reaction,
(DNA)n + dNTP ———————— (DNA)n+1 + PPi (Polymerase)
The addition of one dNTP removes one pyrophosphate from the DNA.
PPi + APS —————————– ATP + SO4-2 (ATP sulfurylase)
ATP + luciferin + O2 ——————— AMP + PPi + oxyluciferin + CO2 + photon (luciferase)
Here the reaction is completed in three steps:
The enzyme polymerase adds the dNTPs to the single-stranded DNA. If the correct complementary base is added, the pyrophosphate is released.
The enzyme sulfurylase converts the PPi into ATP (energy) with the help of the APS (adenosine 5´ phosphosulfate).
The ATP acts as a substrate for the luciferase activity (specifically “firefly luciferase”). With the help of the ATP substrate, the luciferase converts the luciferin into oxyluciferin in the presence of oxygen and the photon of light is released.
Once the correct nucleotide is added, the amount of the light released by the enzymatic reaction is detected by the charged device coupled camera, photodiode, or a photomultiplier tube. This is the basic fundamental of the pyrosequencing setup.
Based on the substrate used in the technique two types of pyrosequencing methods are available: solid-phase pyroseq and liquid-phase pyroseq. We will discuss each type of pyrosequencing in some other articles.
Applications of pyrosequencing:
- The present technique is specifically used in microbial diagnosis, for example, detection of H1N1 during an influenza pandemic.
- Often used in microbial drug resistance studies and analysis.
- It allows accurate and speedy methylation studies.
- Genotyping and mutational studies.
| Advantages | limitations |
| The present method is speedy, robust and automated. It is more accurate than sanger sequencing. Add 500 nucleotides in a single run. | Short read length.Required extensive chemical setup. Works on the principle of chain termination. |
Whole-genome shotgun sequencing:
- Year: 1979
- Scientists: Sanger and Co-workers
- Other Names: – Whole-genome sequencing.
- Automation: Possible.
Yet another modification of Sanger’s chain termination method is the whole-genome shotgun sequencing. However, instead of a single gene or a few base pairs, the present method is powerful enough to sequence the entire genome of an organism.
The principle of the shotgun is the same as Sanger’s method, one additional step of DNA fragmentation allows it to read multiple fragments.
Principle:
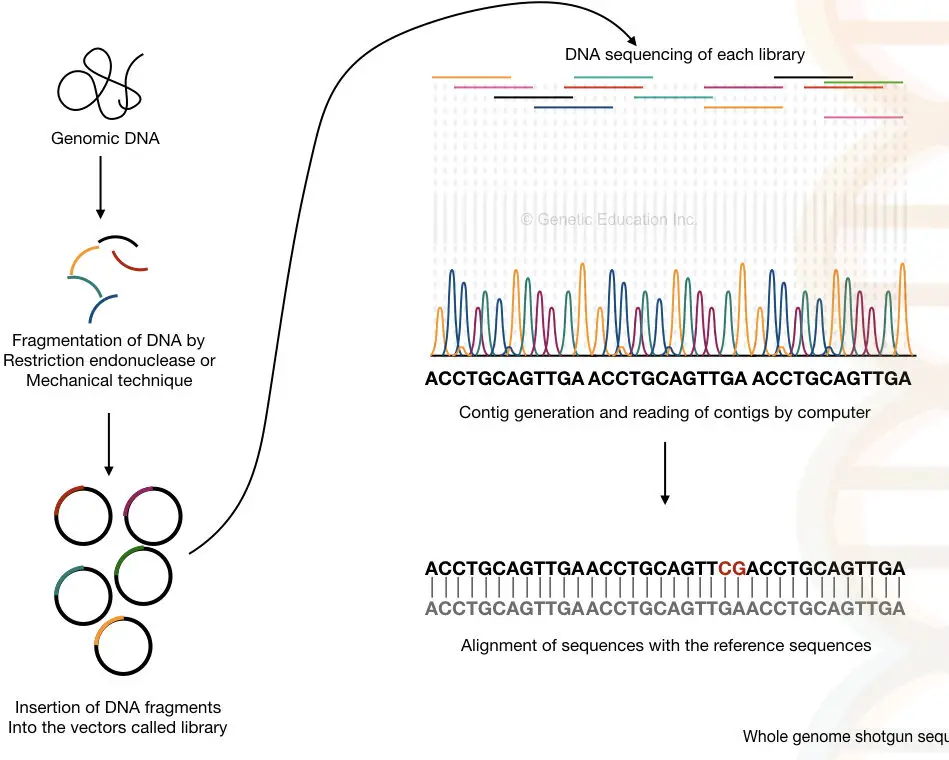
The entire genome of an organism is fragmented with the help of endonuclease enzymes or by mechanical techniques. After that, the smaller fragments of DNA are sequenced individually into the machine.
The computer-based software analyses each and every overlapping fragment and reassembles it to generate the complete sequence of the entire genome.
Process:
The method can be divided into six steps:
- DNA extraction- using any DNA extraction method.
- Fragmentation of DNA- with the help of restriction endonucleases or physical methods.
- Adaptor ligation and library preparation- the fragments are ligated in vectors and an entire library for various vectors is generated.
- Library enrichment- multiple copies of fragments of each library are generated.
- Sequencing- each library is sequenced individually.
- Generation and reading of the contigs- the overlapping fragments called contigs are read by the computer.
Fragmentation is an important process here that generates DNA fragments of 2 to 20Kb which sometimes can be sub-fragments into even smaller sequences.
It is also important to note that the double sequence of each strand helps fill the gaps that remained unsequenced during the process. I have prepared a hypothetical illustration of the process. Take a look.
Do you know?
The shotgun sequencing concept was originally discovered by Sanger F and his colleagues for sequencing the whole genome.

Applications of Shotgun sequencing:
The present method is faster and cheaper than the previous technique. It is one of the three common genome sequencing methods.
The technique becomes more aggressive if any reference sequence is available to align (it can find out the gaps and mutations as fast as possible).
Gene or chromosomal mapping and tedious enzymatic reaction steps are not required in the shotgun sequencing method.
It has the power to sequence the whole genome of an organism (more time is required to sequence the complex genome such as humans and animals).
Nonetheless, it is important to know that the technique becomes feeble if reference genome sequence data is not available.
| Advantages | Limitations |
| Can sequence the entire genome. Accurate and robust than the conventional Sanger technique. Identify gaps and variations in a sequence. | Time-consuming, lengthy and tedious. Need a reference sequence to investigate the results. More error-prone. Require additional steps like fragmentation and library preparation. Overall, more costly. |
“In the year 1981, The genome of cauliflower mosaic virus was sequenced by the shotgun sequencing method.”
Clone by clone sequencing:
- Year: 1980 and 1990
- Scientists: –
- Other Names: –
- Automation: –
For sequencing the whole genome, the clone by clone method is also used instead of whole genome shotgun sequencing. notably, this present method has helped to successfully complete the human genome project.
Principle:
Larger chunks of genomic DNA are sequenced by Sanger’s principle and mapped back by comparing with a pre-prepared chromosome map.
Process:
The present method is more or less similar to the previous method with one additional step. First, instead of smaller fragments, large clumps of DNA fragments are constructed. Through gene mapping, the location of each fragment is noted.
Using the BACs- bacterial artificial chromosome, multiple copies of each fragment are generated for accurate sequencing. In the next step, every copied fragment is fragmented into smaller pieces and inserted into the vectors.
The sequencing is performed as the shotgun and the overlapping fragments are assembled by the computer.
In the last step, the data of the gene or chromosome mapping generated was previously employed to assemble the sequence. The sequences are arranged on each chromosome based on their location.
Applications:
It is applied in the whole genome or chromosome sequencing.
Any gaps in sequencing can be identified easily because the fragment of the DNA is taken from the known locations.
Scientists can work on various chromosomes at once and hence a large number of DNA can be sequenced at the same time.
| Advantages | Limitations |
| More read length. Faster Identify location of sequence variation | Tedious and time-consuming. Requires steps like cloning, fragmentation and digestion. Costlier. Difficult to sequence centromeric and telomeric parts. |
Note: The clone-by-clone sequencing has one major limitation, as cloning of chromosomal parts like telomeres and centromeres is difficult, it can’t accurately sequence them.
As the clone by clone sequencing method was used during the human genome project, it is a part of our history. And hence, its discovery was one of the valuable milestones in the history of genetics.
Interestingly, the shotgun sequencing method evolved from the clone by clone sequencing method.
“During the years 1980 and 1990, the genomes of the nematode worm, C. elegans and the yeast, S. cerevisiae was sequenced using the clone by clone sequencing.”
After reading all of this stuff, you came to know that each technique has one or more serious drawbacks. So you may wonder, is there any platform available that is fast, accurate, reliable, accurate and cost-effective? The answer is “yes”.
Next-generation sequencing is the next level of revolution in sequencing technology.
Next generation sequencing:
- Year: ~2009
- Scientists: – McCooke N (Solexa)
- Other Names: – high throughput sequencing
- Automation: Possible
| Sequencing platform | Read length | High throughput | Accuracy | Run time |
| Next generation sequencing | >100bp (PacBio) | 500bp (PacBio) | 99% | 20 minutes |
As Illumina stated, “it’s an ultra high throughput sequencing technique.” Next-generation sequencing abbreviated as NGS is a revolutionary technique in the field of DNA sequencing.
Due to its high speed, precision and property to sequence the entire genome or gene in a massively parallel fashion, it is employed in microbial genetics, genetic research and even in diagnostic industries.
Principle:
The fragments of DNA or RNA are ligated with adaptors to create genomic libraries which are sequenced in the massively parallel event. The final results are reassembled and analyzed to study the genome.
Steps and Process:
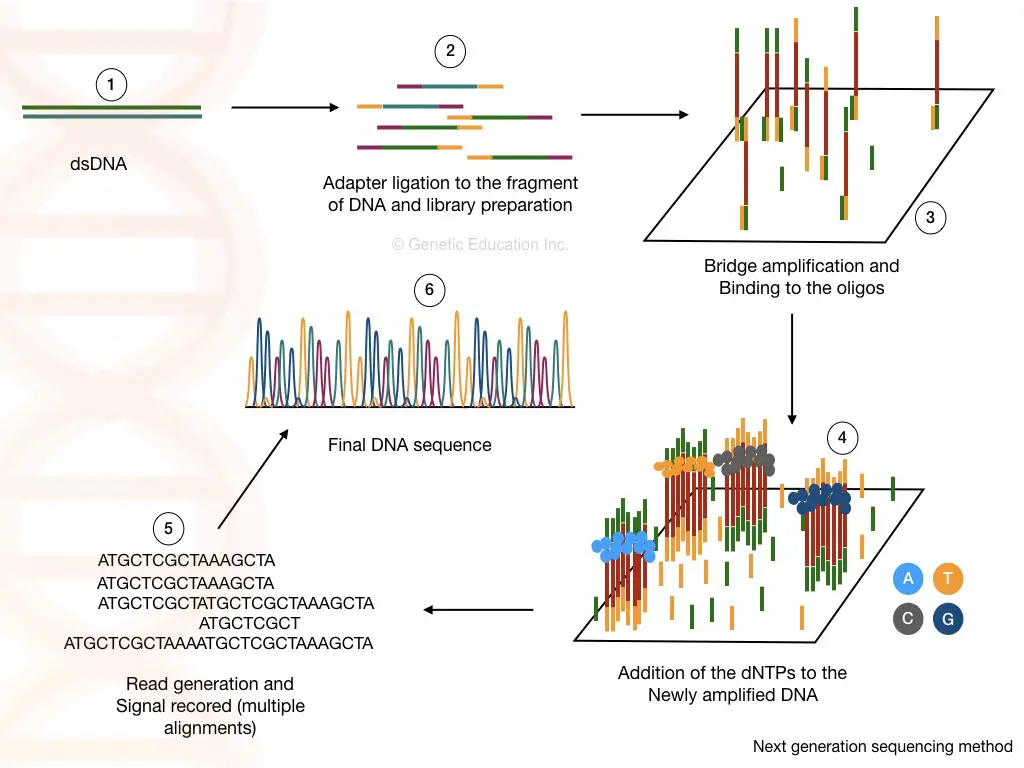
Library preparation:
The library preparation is the combination of two reactions viz, fragmentation and ligation. Using restriction digestion any cDNA or DNA is fragmented into readable pieces. The enzyme ligase joins the fragments with a known sequence.
The known DNA sequences are called the adapters and the process is called adapter ligation. Fragmentation and adaptor ligation generates either a cDNA library or genomic DNA library.
Washing helps to remove unbound fragments, adaptors and other reagents which certainly can influence the sequencing reaction. In the NGS terminology, the process of library preparation is called as tagmentation.
Cluster generation:
What is the role of adapters??
The short oligonucleotide sequences are immobilized on the solid surface which is complementary to our adapter sequences.
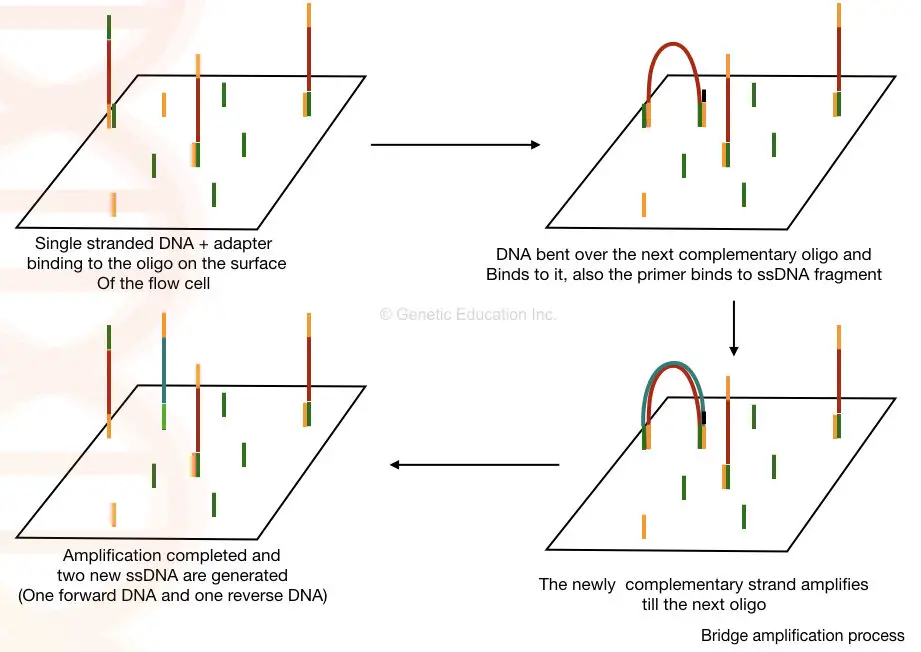
Once the library of our fragmented DNA is loaded into the cell, it is bound with the immobilized oligos on the solid surface. The process of bridge amplification generates clusters of DNA sequences.
Here, in the bridge amplification, the DNA fragments bend over and bind to the next oligo which creates a bridge. A primer binds to this DNA sequence and is amplified vertically.
Two new single-stranded DNA are generated by bridge amplification. See the image below,
Sequencing:
As the polymerase adds the nucleotide into the bridge amplification, the amplification signals are recorded each time. This will generate multiple sequencing databases for the DNA sequence.
Data analysis:
The read generated by the sequencing can be aligned to the reference genome sequence and by doing this we can identify any addition, deletion or variation in the sequence.
Applications:
- Effectively sequence the whole genome of any organism.
- Allow deep sequence analysis.
- Study epigenome.
- Sequence RNA as well.
- Identify novel pathogens, strains, mutations and other variations.
- Study and identify novel and rare genetic variants.
| Advantages | limitations |
| Ultra-high throughput, scalable, precise, speedy and robust. Separate PCR amplification is not required. | Costly. |
Other sequencing methods:
- Single-molecule real-time (RNAP) sequencing
- Single-molecule SMRT(TM) sequencing
- Helioscope(TM) single-molecule sequencing
- DNA nano ball sequencing
- SOLiD sequencing
- Illumina (Solexa) sequencing
- Polony sequencing
- massively parallel signature sequencing (MPSS)
- High throughput sequencing
Applications of DNA sequencing
Sequencing technology has deep penetration in the research and diagnosis industries. Here is the list of some of the important applications.
In medical science, sequencing allows us to study hereditary disorders, a gene or alterations responsible for that. In addition, it can even find novel variants.
Recent trends show that speedy and accurate sequencing can help to study pandemics and epidemics, and finds pathogen and pathogenic variations. For example, techniques like Next-generation sequencing greatly increased screening and diagnosis of COVID-19 in 2020 and 2021.
In forensic science, it is used for parental verification, criminal investigation and identification of individuals through any of the available samples such as hair, nail, blood or tissue.
In agriculture, agri-business and agri-research, such technologies facilitate GMO studies, induced mutagenesis analysis and identification of genomic variations in plant genomes. Thereof scientists can study governing traits.
Accurate chromosome mapping, restriction maps and genome maps can be constructed and analyzed.
Sub-genomic and non-coding genomic entities like non-open reading frames, promoter and enhancer locations can be studied and identified along with coding genes. Other genomic regions like exons, introns, and repetitive portions can also be studied.
Interestingly, whereas it has crucial implications in gene manipulation and gene editing research, novel alterations in nature can be determined.
Techniques like pyrosequencing have great utility in metagenomic analysis and microbiome studies.
Unlike the traditional microbiology process, sequencing equips microbiologists for speedy and accurate microbial identification thereby characterization and classification of microbes. Techniques like 16s rRNA gene sequencing is the best innovative platform in this scenario.
In the process, researchers compare the sequence of any unknown microbe with the available sequence data and find many new things from the genome.
Sequencing revolutionized oncology and cancer research. NGS, for example, changes the field entirely. It can finds cancer-causing genes, alterations, gaps and genomic regions and correlate them to cancer.
Moreover, sequencing also changed the field of evolution. Techniques like molecular phylogeny can be used to construct a DNA sequence-based evolutionary tree. Such analyses are more accurate and precise.
Thereof, an evolutionary map of organisms can be prepared.
Fascinatingly, sequencing helps in studying the asymptomatic high-risk population, prior to the occurrence of disease. And thus preventive steps can be taken earlier.
Do You Know?

Sir Shankar Balasubramanian is the only known Indian scientist who was involved in the development of a sequencing platform. He was the principal investigator in the development of Solexa/ Illumina Next-generation sequencing.
Limitations of DNA sequencing
A sequencing platform is a computer algorithm-based assistive technique that relies on computational data processing. For that, a huge, high-speed supercomputer is required.
Also, several sequences like tandem repeats, repetitive DNA, fragmented genes, and other duplicated regions can not be studied properly. The chance of errors in the pre-sample processing can cause big economical loss, these two are the major limitations of the DNA sequencing methods.
Wrapping up:
Sequencing has infinite scopes in many diverse biological fields. Scientists and medical professionals are looking forward to using it in studying multigenic genetic disorders, and prenatal and pre-implantation genetic studies.
Without the utilization of robust NGS-like platforms, the field of personalized medicines and gene therapies will remain a daydream. Nonetheless, optimizations, advancements and automation are required to reduce cost, time and error rate in the present genetic technology.
Sequencing has immense potential but in recent times, it has, technically, as per my opinion, many limitations. And the biggest problem is that not a universal platform for all analysis is still available.
Source:
- Brown TA. Genomes. 2nd edition. Oxford: Wiley-Liss; 2002. Chapter 6, Sequencing Genomes.
- Heather JM, Chain B. The sequence of sequencers: The history of sequencing DNA. Genomics. 2016;107(1):1-8.
- Lehrach H. DNA sequencing methods in human genetics and disease research. F1000Prime Rep. 2013;5:34. Published 2013 Sep 2.

Which methods have been added since the article and which methods have prevailed and when can you answer this. Thank you very much
what is your question?
Itx amazing article honestly…briefly it provide every content…..
can you solve it plz..Which method scientists adopted for sequencing genome before human genome project, explain in detail..?
We already have! the project was started with the basic Sanger sequencing technique and soon after, replaced by shotgun sequencing technique.
We already have! the project was started with the basic Sanger sequencing technique and soon after, replaced by the shotgun sequencing technique.
Thank you ..
I like how you mentioned that sample preparation is the first step in DNA sequencing. I am interested in DNA sequencing and want to learn more about it. I will definitely keep your information in mind and decide if DNA sequencing is a career path I want to take.
Thank you, Brooklyn Johnson, for appreciation. If you like the content of the article please share it with other friends.
Amazing article!!
Thank you so much Nutricao indeias