“The copy number variation is defined as the alteration that occurs due to change in the number of copies of a gene or genes or DNA due to translocation, deletion, duplication or inversion.”
Or at a chromosomal level, it is defined as,
“Variations in the copy number of chromosome portion due to deletion or duplication.”
Approx. 99% of the human genome is similar to 6 billion bases and around 22,000 to 25,000 different sized genes. The alteration, we knew it as the mutation is the reason new traits and phenotype originate and probably it’s the reason which makes us different.
Change/mutation or alteration are important words for us. We are evolved and become the most advanced species on earth due to changes over time in our genome.
Further to this, the uniqueness of every individual also originated from these mutations and their interactions with the environment.
The mechanism of alteration changed completely when SNP-single nucleotide polymorphism was evolved, stating that change in a single base may also produce alteration and disease susceptible genotypes.
There are millions of SNPs in our genome, perhaps, a major portion of SNPs are non-pathogenic but are involved in the disease.
The mechanism of variation becomes more understandable when the concept of copy number variation (CNV) is postulated recently.
It states that changes in not only the sequence of nucleotides but also its copy number may cause a variable phenotype.
To understand the concept of copy number variation more clearly, we need to learn the composition of the genome.
The genome comprises non-coding intervening sequences of 97% and coding gene sequences of 3% approximately. The non-coding DNA is repetitive in nature and arranged on the telomere and centromeric part of the chromosome.
Note here that the copy number variation comprises not only coding genes but also the entire portion of more than 1kb in size. This means the alteration in the number of copies of the entire region of more than 1kb may have a few or a few hundred genes that exactly the CNVs are! So keep in mind that it’s the entire portion of chromosomes.
But you wonder how we can detect it?
In the present article, we will learn about copy number variation and its detection methods. Also, we will understand how the quantification is done and what is the role of copy number variation in evolution.
Interesting read: Differences between Genotyping vs Sequencing.
Key Topics:
What is copy number variation?
Before we go into more technical understanding, let us first understand the mechanism of copy number variation with one example. We all heard about Huntington’s disease, right!
It’s a triplet repeat expansion disease. Here The short triplet repeat on the chromosome expands abnormally in the patient. This means that If a normal person has 23 CAG repeats, the abnormal has more than this. And as the number of repeats increases, the severity of the disease increases.
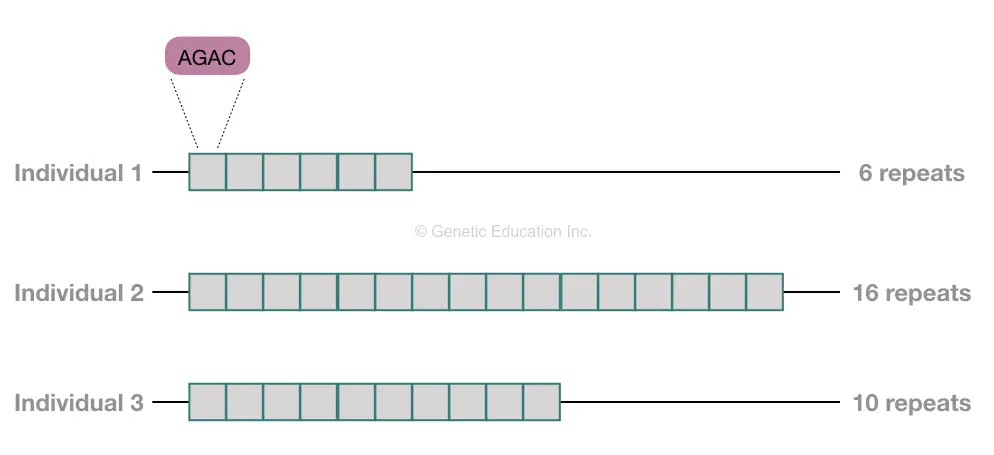
What it means, duplication of genetic content repeatedly.
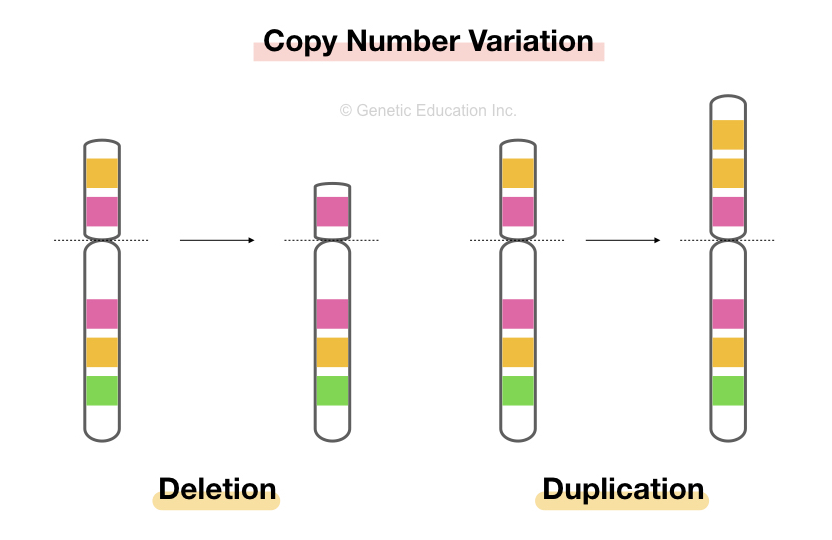
As we discussed above, the region of more than 1kb is taken into account for CNVs study. At a chromosomal level, deletion and duplication cause a major change in copy numbers.
Simply put, It is just a change in the number of nucleotide sequences between two organisms. We can also understand it by taking an example of Down syndrome in which the whole chromosome (21) got duplicated which increases the number of gene copies present on chromosome 21.
Related article: Trinucleotide repeat expansion disorders.
Definition of copy number variation:
Variation in the number of more than 1kb regions of the genome in different organisms due to either chromosomal deletion or duplication is known as copy number variation and abbreviated as CNVs.
In 2006, the international HapMap project was launched with the aim to find out possible copy number variations in different ethnic populations. Subjects of African, Asian and European descents were selected to make a database of CNVs. Redon et al, involved in the HapMap project, extracted several crucial information from it, as stated,
12% of the human genome region is prone to copy number variation which is 360Mb in size. This means it is hypervariable.
1447 different copy number variation sites were reported in their study. The major cause of CNVs is chromosomal duplications and deletions.
40 different CNVs are directly linked to the disease.
The variability of CVNs in different persons varies from 6% to 19%.
More recent studies after these findings suggest that 0.5% of the genomic portion with roughly 600 to 900 different CNVs are different between two people.
Genes are supposed to be present in a pair on each chromosome, said traditional research but due to change in copy numbers, it alters!
In trisomies and monosomies, three copies and single copies of genes are present instead of two, respectively.
Causes of CNVs:
SNPs may follow Mendelian inheritance or non-mendelian or sometimes occur randomly. But till now no clear reason or cause for the category copy number variation is formulated.
However, a brief understanding suggested that change in copy numbers may occur due to the interaction of genome and environment. And that is the reason some harsh environments are so lethal for some individuals.
The mechanism of non-disjunction which occurs randomly during cell division falsifies the belief, at least in the case of aneuploidies. The possibility of the involvement of environmental factors can’t be ruled out completely.
Genetically several CVNs are inherited and follow a distinct pattern while the rest are undefined. Gradual increase and decrease in gene copies influence the related phenotype adversely.
Techniques to detect copy number variations:
Karyotyping:
One of the most traditional methods to evaluate the copy number variations visually is the karyotyping method. It’s a cell culture-based method to study the metaphase chromosomes.
The present method detects copy number variations like larger deletion, duplications and changes in the number of chromosomes.
Read more: Karyotyping: Definition, Steps, Procedure and Applications.
FISH:
Known as Fluorescence in situ hybridization is a technique to screen the deletions and duplications more precisely than karyotyping.
In the FISH technique, the fluorescent probes are used to hybridize with the target location to map the deletion or duplication on the chromosome.
Probes bind on the metaphase chromosome and give signals. Though the present technique relies upon fluorescence chemistry, it is more accurate and efficient than the conventional technique.
Read our article on FISH: introduction of fluorescence in situ hybridization.
Array CGH:
Array CGH or the microarray is another DNA hybridization-based method on which thousands of copy number variations can be screened on a single slide. The fluoro-labeled probes are immobilized on the glass slide instead. And the sample of control and sample DNA is allowed to hybridize on it.
The Hybridization pattern of deletion and duplication on variation chromosomes can be encountered in a simple experiment.
This method is advantageous over the conventional karyotyping and FISH, as it is fast, and can screen thousands of copy number variations at once.
Besides these cytogenetic methods, next-generation sequencing and PCR are also used in the CNV studies. Next-generation sequencing is a high throughput sequencing method that can sequence the whole genome and can determine the number of the whole genome.
Furthermore, it is more advanced than other methods as we can identify new copy number variations as well. Read more: Genome on a chip- DNA microarray.
DNA sequencing:
The present method is yet powerful enough to identify the variable locus from the genome. It can read the sequence as well as identify deletion or duplications. The sequencing method is powerful enough to find out new variations.
PCR:
A machine known as a thermocycler can perform the reaction known as a polymerase chain reaction that can detect polymorphic sites in DNA.
An optimized version of it known as real-time PCR can even detect the number of copies of a gene or polymorphic locus.
CNVs of the disease like Fragile X and Huntington’s can be encountered using quantitative PCR. Read our whole article on PCR: Polymerase chain reaction.
CNVs and evolution:
The mechanism of copy number variations is not only involved in disease development but also helps us to evolve, as per the recent findings.
Scientists have studied the genome of humans and chimpanzees to relate the CVNs which suggests that though a major portion of CNVs of both genomes is similar, a few have just evolved recently, and also have positive selection effects.
Furthermore, several copy number changes are also reported just recently in the higher species-specific conserved regions of humans that also favor a positive effect of evolution.
Nonetheless, we have less published data that conclude the role of CNVs in evolution, unlike the role of transposons in evolution. But studies strongly indicated that inversion, deletion and duplication of genetic content might have played a pivotal role in human evolution.
Conclusion:
The field copy number variation studies are new to us. There are so many links that are still missing yet the role of CNV is significant in our genome and the development of the disease.
For instance, the severity of the disease in case of some duplication depends on how many portions of the chromosome got duplicated. It indicates that CNV is one of the critical genetic factors much like the SNPs.
DNA sequencing and ArrayCHG are yet not powerful enough to detect all the CNVs that are the biggest limitation of it. Still, scientists are trying hard to develop new tools and techniques to study CNVs.
Sources:
Redon, R., et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006) doi:10.1038/nature05329
Stefansson, H., et al. A common inversion under selection in the human genome. Nature Genetics 37, 129–137 (2005) doi:10.1038/ng1508