
“Learn about the concept of trinucleotides, trinucleotide repeats and trinucleotide expansion disorders. Explore the mechanisms and types of trinucleotide expansion disorders in this article.”
Nucleotides are the building blocks and basic unit of the DNA. It’s a composition of sugar, phosphate and a nitrogenous base. Functionally, it provides stability to DNA structure.
The long chain of nucleotides, which we refer to as ‘DNA sequences’ are the clouds of nucleotide sequences. A specific sequence that synthesizes a protein is known as a “gene.” However, other non-specific or unknown sequences are also there in our genome and likely to work in epigenetic regulation.
Nucleotides are the constituents of the genetic code– that triplet codons, we explain them as ‘CAA, GGT, GTG or TCT, etc’ translate into amino acids. Interestingly, the number of triplets for a gene or non-gene sequence also matters!
What does that mean?
A particular type of trinucleotide, if exceeds its original number, may result in disease conditions. Such conditions are broadly categorized into trinucleotide expansion disorders.
In this article, I will explain the concept of Trinucleotide Expansion Disorders and related topics. We will explore the concept of triplet repeats and understand the mechanism behind it. At the very end, I will give you the list of disorders.
I worked for years in a genetic lab and during that period I tested thousands of samples including the samples having triplet repeat expansion disorders. I researched this topic during that period and gained substantial topical authority.
I also have published one review article on this topic as well. So I’m the better person to explain this topic.
Stay tuned.
Key Topics:
What is trinucleotide repeat expansion?
Trinucleotides are the group of three nucleotide bases, for instance, CAG, GGG or GAT. When these trinucleotides occur one after another in the genome with a specific number, it’s known as trinucleotide repeats.
For example,
| Repeat | No. |
| CAG CAG CAG CAG CAG CAG CAG | 7 |
| GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT GCT | 12 |
As you can see in the example, the number of CAG repeats in the first sequence is 7 while the number of GCT repeats in the second sequence is 12. These examples finely define the concept of trinucleotide repeats.
So, conclusively, the trinucleotide repeats are the number of times any three nucleotides repetitively present in a sequence or a gene. Interestingly, when these numbers increase unwantedly, it’s known as trinucleotide expansion or triplet repeat expansion. And that my friends, creates a big health problem.
Let’s understand the concept more keenly.
Repetitive DNA, though, doesn’t have any substantial functionality in our genome but still plays a crucial role. Such repetitive DNA sequences are present in telomeres, in non-coding/ heterochromatin regions and even in coding genes.
Interestingly, shorter repeats of 1 to 6 nucleotides (STRs) and longer repeats of up to 100 nucleotides (VNTRs) have been used in genetic testing for years. So the concept of repetitive DNA wasn’t new to genetics.
However, the role of trinucleotide repeat expansions in disease origin was just introduced in the early 1990s. When a specific type of trinucleotide expands to its original length in a mutant gene, it results in a disease condition.
For example, the CGG repeats present on the 5’ UTR of the FMR1 gene have 6 to 50 (approximately) normal repeats, however, when the number of CGG repeats increases from 200 to 4000 times, it results in Fragile X syndrome.
Interestingly, trinucleotide expansion occurs within coding as well as within the non-coding sequences as well. So we can categorize it neither in genetic nor in epigenetic alterations, ideally.
It occurs within the gene (in a coding region), for instance— Huntington’s disease (CAG expansion) and in the non-coding parts, for instance— Myotonic dystrophy 1.
This special class of mutation follows the mechanism of genetic anticipation, meaning, as the expansion is transmitted to the offspring, the number of repeats increases, and so does the disease severity!
Trinucleotide repeat expansion condition is also known as triplet repeat expansion or TNRs.
In 1991, Stephen Warren explained the unstable nature of the CGG repeats and their role in the Fragile X syndrome. Later on, in 1993, Gusella J et al. demonstrated abnormal CAG repeats from the HTT gene and its role in Huntington’s disease. Here is the short history of the TNRs.
| Year | Source | TNR | Gene | Disease |
| 1991 | Stephen Warren | CGG | FMR1 | Fragile X syndrome |
| 1993 | James Gusella et al. | CAG | HTT | Huntington’s disease |
| 1992 | Charles Thornton et al. | CTG | DMPK | Myotonic dystrophy |
| 1993 | Stefan Pulst et al. | CAG | ATXN2 | SCA2 |
| 1994 | Huda Zoghbi and Harry Orr | CAG | ATXN3 | SCA |
How does Trinucleotide repeat expansion occur?
Now, in this section, we will understand a few mechanisms behind the expansion of triplet repeats, in general. Although the clear mechanism behind the repeat expansion is still unclear, abnormalities in some pathways have been designated as repeat expansion culprits.
Replication slippage is one such condition that can potentially result in trinucleotide repeat expansion. Note that abnormal repeat expansion occurs in both dividing and nondividing cells.
Replication slippage:
Replication slippage extensively occurs in the repetitive DNA regions, i.e. in trinucleotide repeat regions as well. During the normal replication cycle, the double-stranded DNA separates into two single-stranded DNA and provides a template for replication.
After that, the DNA polymerase uses the primer DNA junction, initiates nucleotide addition and synthesizes a new DNA strand. Oftentimes, that does not occur in highly repetitive regions like CAG, CTG or CGG right sequences.
These trinucleotide repeat regions are highly prone to replication slippage and result in abnormal repeat expansion. Now, what happens here?
Sometimes, this repetitive place forms a temporary hairpin or loop-like structure by self-base-pairing in the template DNA, detaches temporarily and misses the synthesis process by DNA polymerase.
Resultantly, when both the strands re-join, they can’t align correctly. To overcome the present situation, replication either adds or removes extra nucleotides in the process. If it adds new nucleotides, it causes repeat expansion.
If it removes the nucleotides, it causes repeat loss. In the present case, replication slippage results in the addition of nucleotides and expansion of triplet repeats.
As the replication progresses, new loops are formed and newer nucleotides are continuously added to the growing DNA strand to overcome mismatch. Thus, after each cell division, the triplet further expands.
Replication slippage is well-established for trinucleotide expansions in Fragile X syndrome and Huntington’s disease.
Yet another mechanism referring to an error in Transcription-mediated DNA repair is also explained in some literature. It’s commonly observed in non-dividing cells.
Transcription-mediated DNA repair:
The present hypothesis explains that the gene-specific trinucleotide expansion may also occur during the transcription or transcriptional repair stage. A DNA repair pathway dedicatedly allocated to correct errors in transcriptionally active genes.
When RNA polymerase comes across the trinucleotide repeat regions, it considers it as an abnormality and triggers the DNA repair pathway. Now what the repair mechanism does is that it either adds new nucleotides or removes some to avoid mispairing.
In case it adds nucleotide, it results in the expansion of that particular triplet repeat. Here also, it follows the mechanism of genetic anticipation and after every round of replication or generations, the number of repeats gradually increases.
And
So does the disease severity!
These are the two common theories likely to contribute to triplet repeat expansion.
Genetic Anticipation in Trinucleotide Expansion Disorders:
Now, after discussing the mechanisms behind the repeat expansion, we also have to understand the anticipation mechanism to better understand the disease condition and severity.
In my explanation, genetic anticipation is a kind of disease pattern in which the severity of the disease increases and the age of disease onset decreases in successive generations or offspring.
For instance, suppose one of the parents from a family has 40 CAG repeats and is diagnosed with Huntington’s disease penetration at the age of 35. Now 50% of their children would have Huntington’s disease with even more CAG repeats (>40), severe disease symptoms and early disease onset.
This pattern continues to consecutive generations as well. Check out the hypothetical pattern chart here.
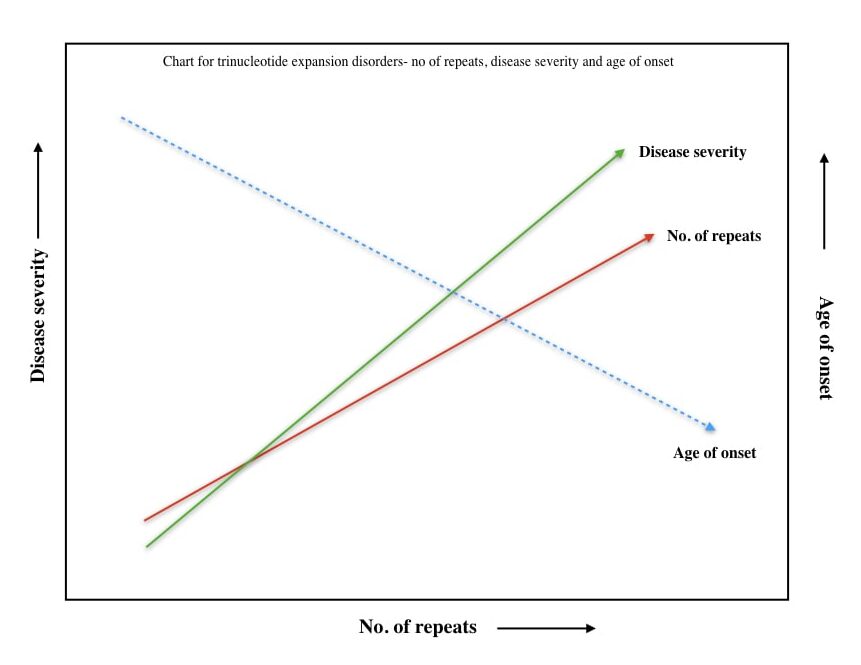
So anticipation begins with the very first round of either replication strand slippage or transcriptional-mediated DNA repair and will continue after each round of cell division.
Thus, more and more repeats are added after each cell cycle and/or generation. The consequences of genetic anticipation are listed here,
- Expansion of the trinucleotide repeats after every cell cycle.
- Increase in disease penetration.
- More and more new symptoms appear as the progression affects more new systems.
- The disease severity increases after each generation.
- The age of disease onset decreases after each generation.
List of Trinucleotide Expansion Disorders:
Here I’m giving you the list of disorders that occurred by the triplet repeat expansion.
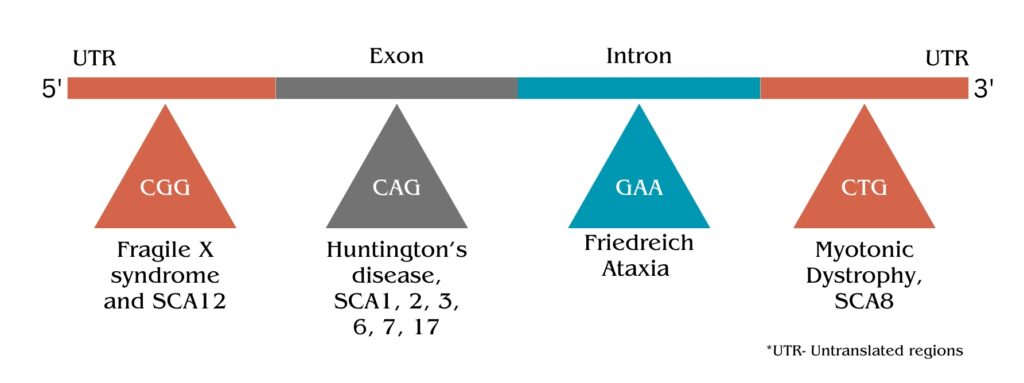
Huntington’s Disease (HD):
- Repeat: CAG
- Gene: HTT
- Normal Range: 10-35 repeats
- Disease Range: Typically 36 or more repeats
Myotonic Dystrophy Type 1 (DM1):
- Repeat: CTG
- Gene: DMPK
- Normal Range: 5-37 repeats
- Disease Range: 50 or more repeats
Fragile X Syndrome:
- Repeat: CGG
- Gene: FMR1
- Normal Range: Up to about 45 repeats
- Pre-mutation: 55-200 repeats (associated with increased risk of fragile X-associated disorders)
- Full Mutation: Over 200 repeats (causes fragile X syndrome)
Spinocerebellar Ataxia Type 1 (SCA1):
- Repeat: CAG
- Gene: ATXN1
- Normal Range: ~6-44 repeats
- Disease Range: 45 or more repeats
Spinocerebellar Ataxia Type 2 (SCA2):
- Repeat: CAG
- Gene: ATXN2
- Normal Range: Up to 31 repeats
- Disease Range: Over 32 repeats
Spinocerebellar Ataxia Type 3 (SCA3), also known as Machado-Joseph Disease (MJD):
- Repeat: CAG
- Gene: ATXN3
- Normal Range: 12-44 repeats
- Disease Range: 45 or more repeats
Spinocerebellar Ataxia Type 6 (SCA6):
- Repeat: CAG
- Gene: CACNA1A
- Normal Range: 4-18 repeats
- Disease Range: Over 19 repeats
Friedreich’s Ataxia:
- Repeat: GAA
- Gene: FXN
- Normal Range: 5-30 repeats
- Disease Range: Over 200 repeats
Dentatorubral-pallidoluysian Atrophy (DRPLA):
- Repeat: CAG
- Gene: ATN1
- Normal Range: 6-35 repeats
- Disease Range: Over 49 repeats
Spinocerebellar Ataxia Type 7 (SCA7):
- Repeat: CAG
- Gene: ATXN7
- Normal Range: Up to 17 repeats
- Disease Range: Over 36 repeats
Haw River Syndrome (HRS):
- Repeat: CAG
- Gene: TBP
- Normal Range: Up to 35 repeats
- Disease Range: 48 or more repeats
Oculopharyngeal Muscular Dystrophy (OPMD):
- Repeat: GCG
- Gene: PABPN1
- Normal Range: Up to 17 repeats
- Disease Range: 10 or more repeats
Spinocerebellar Ataxia Type 8 (SCA8):
- Repeat: CTG
- Gene: The gene responsible for SCA8 is currently unknown, but the repeat expansion is located on chromosome 13.
- Normal Range: Up to 30 repeats
- Disease Range: Over 110 repeats
Spinocerebellar Ataxia Type 12 (SCA12):
- Repeat: CAG
- Gene: PPP2R2B
- Normal Range: Up to 32 repeats
- Disease Range: Over 66 repeats
Summary
| Sr No. | Disorder | Repeat | Gene | Normal | Abnormal |
| 1 | Huntington’s Disease (HD) | CAG | HTT | 10-35 | >36 |
| 2 | Myotonic Dystrophy Type 1 (DM1) | CTG | DMPK | 5-37 | >50 |
| 3 | Fragile X Syndrome | CGG | FMR1 | <45 | >200 (full mutation) |
| 4 | Spinocerebellar Ataxia Type 1 (SCA1): | CAG | ATXN1 | 6-44 | >45 |
| 5 | Spinocerebellar Ataxia Type 2 (SCA2): | CAG | ATXN2 | <31 | >32 |
| 6 | Spinocerebellar Ataxia Type 3 (SCA3) | CAG | ATXN3 | 12-44 | >45 |
| 7 | Spinocerebellar Ataxia Type 6 (SCA6) | CAG | CACNA1A | 4-18 | >18 |
| 8 | Friedreich’s Ataxia | GAA | FXN | 5-30 | >200 |
| 9 | Dentatorubral-Pallidoluysian Atrophy (DRPLA) | CAG | ATN1 | 6-35 | >49 |
| 10 | Spinocerebellar Ataxia Type 7 (SCA7) | CAG | ATXN7 | <17 | >36 |
| 11 | Spinocerebellar Ataxia Type 8 (SCA8) | CTG | Unknown | Up to 30 | >110 |
| 12 | Spinocerebellar Ataxia Type 12 (SCA12) | CAG | PPP2R2B | <32 | >66 |
| 13 | Haw River Syndrome (HRS) | CAG | TBP | <35 | >48 |
| 14 | Oculopharyngeal Muscular Dystrophy (OPMD) | GCG | PABPN1 | <17 | >10 |
| 15 | FRAX-E | GCC | AFF2 | 4-39 | >200 |
| 16 | FRDA | GAA | FXN | 5-30 | 70-1000 |
Wrapping up:
In the previous version of this article, I explained Huntington’s disease and Fragile X syndrome. But now I realize that it doesn’t fit in this article. So I removed it but I will add it to some new article.
Triplex repeat expansion disorders are a special class of genetic diseases having a complex clinical foundation. I, with my fellow scientists, conducted some research on this topic. As I mentioned above, I published a review article on this topic. The link is here: Neurodegenerative Triplex Expansion Disorders.
If you would like to learn more about this topic, you can go and read that article. Till then bookmark this page and share this article with your friends. In the upcoming articles of this series, we will explore more about this topic.
Sources:
Viguera, E et al. “Replication slippage involves DNA polymerase pausing and dissociation.” The EMBO journal vol. 20,10 (2001): 2587-95. doi:10.1093/emboj/20.10.2587.
Gusella, J., Wexler, N., Conneally, P. et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 (1983). https://doi.org/10.1038/306234a0.
Thornton CA. Myotonic dystrophy. Neurol Clin. 2014 Aug;32(3):705-19, viii. doi: 10.1016/j.ncl.2014.04.011. Epub 2014 Jun 6. PMID: 25037086; PMCID: PMC4105852.
Pulst SM. Spinocerebellar Ataxia Type 2. 1998 Oct 23 [Updated 2019 Feb 14]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1275/.

superb…
healthy content…
keep it up…
suggestions- whatever you include, just cover it completely, as you cover huntington disease very deppld and you leaved fragile x with some brief information. so take one thing at a one time and do it completely or if you focusing on the general part like triple repeat expansion, then focus on each and every category equally.
impressed, your hard work is seen in this article.
great information.
I will follow your suggestion