“Did you know that nearly 10 disorders are caused by CAG repeat expansion among which 6 are SCA? learn about the CAG repeat expansion disorders in this article.”
In the previous article of this series, we introduced the concept of trinucleotides and expansion disorders. We also learned the mechanisms behind the triplet repeat expansion and anticipation.
Now, moving in this article (Trinucleotide repeat expansion disorders 102) we will discuss particularly the CAG repeat expansion and associated disorders.
Stay tuned.
Key Topics:
CAG repeat expansion:
50 different genetic disorders have a clear foundation of trinucleotide expansion disorders, however, among which the majority of disorders are caused by the expansion of a CAG repeats only.
CAG repeat is a long chain of Cytosine, Adenine and Guanine, and encodes for Glutamine amino acid. It is located in the coding part of a gene. Glutamine is denoted as Q. So the present condition is often known as polyglutamine disorder or Poly-Q disorders.
Including Huntington’s disease, SCA type 1, 2, 3, 6, 7 and 17, spinal and bulbar muscular atrophy and DRPLA are caused by the CAG repeat expansion. Each disorder, its inheritance pattern, repeat expansion and description are given here.
CAG repeat expansion disorders:
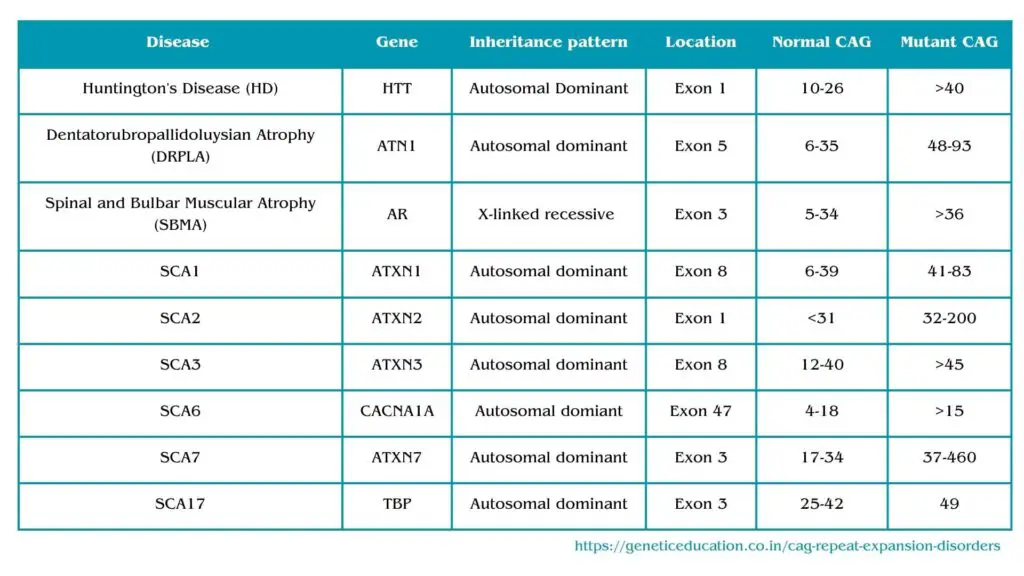
Huntington’s Disease (HD):
Huntington’s disease causes neurodegenerative, motor and cognitive malfunction in the patient.
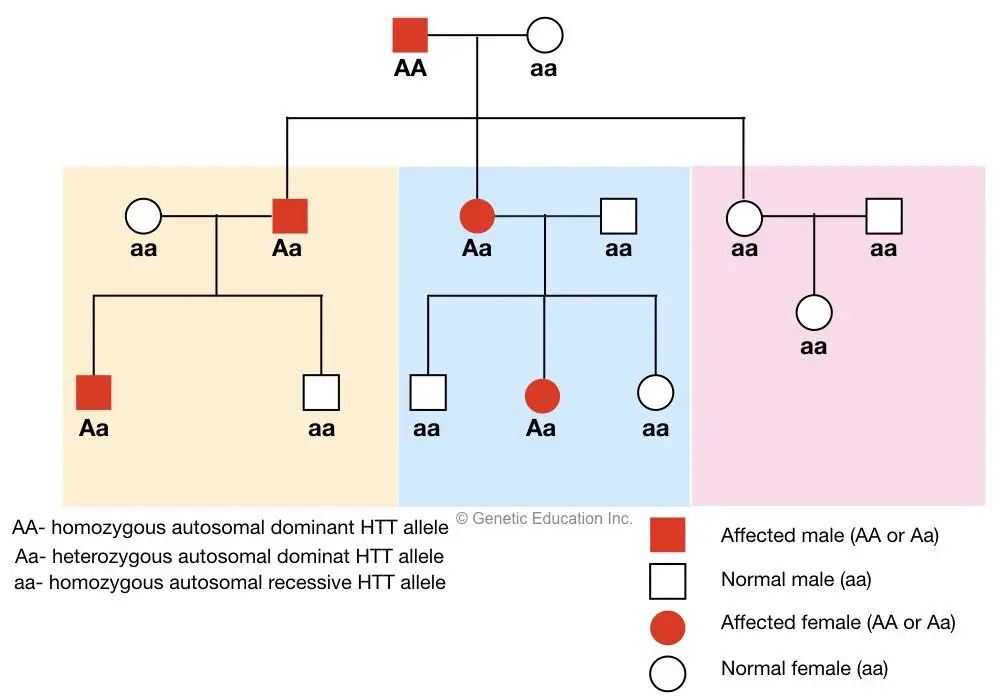
- Inheritance Pattern: Huntington’s disease (HD) follows an autosomal dominant inheritance pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: HTT or huntingtin gene.
- Repeat Type: CAG trinucleotide repeat expansion.
- Number of Repeats:
- Normal Range: In unaffected individuals, the HTT gene typically contains 10 to 26 CAG repeats.
- Intermediate: 27 to 35
- Reduced Penetrance: 36 to 39
- Disease Range: In individuals with Huntington’s disease, the gene typically contains 40 or more CAG repeats in the mutant allele.
- Description:
- The present condition is a neurodegenerative disorder. It occurs typically between the ages of 30 and 50 (adulthood).
- With normal CAG numbers normal huntingtin protein is produced, however, the expanded CAG repeats in the HTT gene lead to the production of a mutant huntingtin protein, which accumulates in neurons and disrupts their function.
- Symptoms of HD include involuntary movements (chorea), motor dysfunction, mood disturbances, cognitive decline, and psychiatric symptoms.
- As the disease progresses, the age of onset decreases in the next generations.
- Individuals with HD often require increasing levels of care and support, and it ultimately leads to severe disability and a shortened lifespan.
If you would like to learn more about this topic, I have written the entire article on this topic. You can read it here: How is Huntigton’s disease inherited?
Dentatorubropallidoluysian Atrophy (DRPLA):
DRPL is associated with a wide range of neurological conditions and thus is also a type of neurodegenerative disorder.
- Inheritance Pattern: Dentatorubropallidoluysian Atrophy (DRPLA) is inherited in an autosomal dominant pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: ATN1 (Atrophin-1) gene.
- Repeat Type: Expansion of CAG repeats.
- Number of Repeats:
- Normal Range: In unaffected individuals, the ATN1 gene typically contains 6 to 35 CAG repeats.
- Intermediate: 35 to 47.
- Disease Range: In individuals with DRPLA, the gene typically contains 48 to 93 CAG repeats in the mutant allele.
- Description:
- Dentatorubropallidoluysian Atrophy (DRPLA) is a rare genetic disorder characterized by a wide range of neurological problems. However, it has a wide variety of clinical manifestations which can also vary among affected patients.
- Much like Huntington’s disease, the expanded CAG repeats within the ATN1 gene lead to the production of an abnormal atrophin-1 protein, which accumulates in neurons and interferes with their function.
- Symptoms of DRPLA can include ataxia (problems with coordination), chorea (involuntary movements), dystonia (muscle contractions), and seizures. In some severe conditions, cognitive problems, psychiatric symptoms or psychosis are also reported.
- Although the age of onset highly depends on the CAG repeat expansion, it may vary also. Some individuals experience symptoms in childhood, while others may not experience symptoms until adulthood.
Spinal and Bulbar Muscular Atrophy (SBMA):
Spinal and Bulbar Muscular Atrophy (SBMA), also known as Kennedy’s Disease, is a rare genetic disorder that primarily affects males. It is characterized by a gradually progressive neuromuscular disorder.
- Inheritance Pattern: Spinal and Bulbar Muscular Atrophy is inherited in an X-linked recessive pattern. This means that the gene associated with SBMA is located on the X chromosome, and the disorder primarily affects males. Carrier females usually do not show symptoms but can pass the mutation to their sons.
- Gene: Androgen Receptor (AR) gene.
- Repeat Type: SBMA is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the AR gene typically contains 5 to 34 CAG repeats.
- Disease Range: In individuals with SBMA, there is an abnormal expansion of CAG repeats, resulting in more than 36 to 37 repeats in the mutant allele.
- Description:
- The expanded CAG repeats within the AR gene lead to the production of a mutant androgen receptor protein, which accumulates in motor neurons and interferes with their function.
- Symptoms of SBMA may include muscle weakness, atrophy (wasting), and fasciculations (muscle twitching). These symptoms typically begin in adulthood and progress slowly.
- In addition to muscle-related symptoms, SBMA can also affect the bulbar muscles involved in speech and swallowing, leading to speech difficulties and dysphagia (difficulty swallowing).
- Although SBMA primarily affects males, female carriers of the mutated gene may experience mild symptoms or be asymptomatic. Symptoms in carrier females are usually less severe than in affected males.
Spinocerebellar Ataxia Type 1 (SCA1):
Spinocerebellar Ataxia Type 1 (SCA1) is a rare genetic disorder characterized by progressive ataxia, which involves problems with coordination and balance. Put simply, it’s a progressive movement and balance problem.
- Inheritance Pattern: Spinocerebellar Ataxia Type 1 (SCA1) is inherited in an autosomal dominant pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: ATXN1 gene.
- Repeat Type: CAG trinucleotide expansion.
- Number of Repeats:
- Normal Range: In unaffected individuals, the ATXN1 gene typically contains 6 to 39 CAG repeats.
- Disease Range: In individuals with SCA1, the gene typically contains 41 to 83 or more CAG repeats in the mutant allele.
Spinocerebellar Ataxia Type 2 (SCA2):
- Inheritance Pattern: Spinocerebellar Ataxia Type 2 (SCA2) is inherited in an autosomal dominant pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: ATXN2 gene.
- Repeat Type: SCA2 is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the ATXN2 gene typically contains up to 31 CAG repeats.
- Disease Range: In individuals with SCA2, the gene typically contains over 32 CAG repeats and up to 200 in the mutant allele.
Spinocerebellar Ataxia Type 3 (SCA3), Machado-Joseph Disease (MJD):
- Inheritance Pattern: Spinocerebellar Ataxia Type 3 (SCA3) follows an autosomal dominant inheritance pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: ATXN3 gene.
- Repeat Type: SCA3 is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the ATXN3 gene typically contains 12 to 40 (or 44 some literature suggests) CAG repeats.
- Disease Range: In individuals with SCA3, the gene typically contains >45 CAG repeats in the mutant allele.
Spinocerebellar Ataxia Type 6 (SCA6):
- Inheritance Pattern: Spinocerebellar Ataxia Type 6 (SCA6) follows an autosomal dominant inheritance pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: CACNA1A gene.
- Repeat Type: SCA6 is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the CACNA1A gene typically contains 4 to 18 CAG repeats.
- Disease Range: affected individuals may carry more than 19 CAG repeats in the CACNA1A gene.
- Description:
- In the present condition, the function of a cerebellar neuron is affected by the mutant CACNA1A gene encoded with an abnormal calcium channel protein.
- Individuals with SCA6 may experience a range of symptoms much like the other types of SCA. Notedly, symptoms typically begin in adulthood.
Spinocerebellar Ataxia Type 7 (SCA7):
- Inheritance Pattern: Spinocerebellar Ataxia Type 7 (SCA7) is inherited in an autosomal dominant pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: ATXN7 gene.
- Repeat Type: SCA7 is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the ATXN7 gene typically contains up to 17-34 CAG repeats.
- Disease Range: In individuals with SCA7, the gene typically contains more than 37 to 460 CAG repeats in the mutant allele.
Spinocerebellar Ataxia Type 17 (SCA17):
- Inheritance Pattern: Spinocerebellar Ataxia Type 17 (SCA17) is inherited in an autosomal dominant pattern. This means that if one parent carries the mutant gene, there is a 50% chance of passing the disorder to their offspring.
- Gene: TBP (TATA-binding protein) gene.
- Repeat Type: SCA17 is caused by a genetic mutation involving the expansion of a CAG trinucleotide repeat.
- Number of Repeats:
- Normal Range: In unaffected individuals, the TBP gene typically contains 25 to 42 CAG repeats.
- Disease Range: In individuals with SCA17, the gene typically contains more than 49 CAG repeats in the mutant allele.
Description of ataxia types:
- The expanded CAG repeats within the gene lead to the production of mutant ataxin proteins, accumulating in neurons and interfering with their function.
- Incoordination, muscle stiffness, slurred speech, and difficulty swallowing are common symptoms reported in patients with various types of SCAs.
- Note that the symptoms may appear aggressively in adulthood. Severe conditions lead to increasing disabilities.
Currently, no cure is available for any of the CAG repeat expansion disorders. However, Genetic testing can determine the expansion of CAG in the patient and also help predict the likelihood of disease inheritance.
Related article: 100 Common Genetic Disorders.
Wrapping:
In conclusion, the expansion of CAG, disease symptoms, and age of onset vary among different conditions and genes. However, it is largely known as PolyQ disorder and causes neurodegenerative problems.
As said in the previous article, you can read my peer-reviewed article to gain more clarity on the present topic. I hope you like this article. Please share it and bookmark the page.
P.S. Review article on Trinucleotide Expansion Disorders.
Sources:
Bouzid, Fatima Zahra et al. “Spinocerebellar ataxia Type 7: clinical and genetic study of a new Moroccan family (case report).” The Pan African medical journal vol. 38 162. 12 Feb. 2021, doi:10.11604/pamj.2021.38.162.27262.
Prades S PhD, Melo de Gusmao C MD, Grimaldi S MD, et al. DRPLA. 1999 Aug 6 [Updated 2023 Sep 21]. In: Adam MP, Mirza GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1491/.