“Learn about the FASTA file, what it is, how to obtain it and use it in various genetic experiments and analysis in this article.”
FASTA is a topic covered in bioinformatics but also has significant importance in genetic research as it contains the sequence of a particular gene or protein, so it becomes equally important for the geneticists to know what the FASTA file is and how to extract and use it.
We are dividing this article into three parts: The first part will focus on what a FASTA sequence is and its format; the second portion will show the step-wise procedure to extract the FASTA sequence from the NCBI database; and in the last part, we will focus on how we can use the FASTA sequence.
Stay tuned.
Key Topics:
What is the FASTA sequence?
The FASTA sequence format is a text-based representation format that uses single-letter codes to represent base pairs or amino acids in either nucleotide or peptide sequences.
An example of a FASTA sequence is as follows:
This is the FASTA sequence of the SRY gene present on Chromosome Y in Homo sapiens.

This is the FASTA sequence of the urease protein in Arabidopsis thaliana.

Now, after seeing the examples of the FASTA sequence, let us understand the format of the FASTA sequence.
In FASTA format, a sequence consists of lines of sequence data after a single-line description. The greater-than (“>”) symbol in the first column separates the description line from the sequence data. It is advised that text lines be no more than 80 characters long.
Occasionally, the FASTA format is also called the “Pearson” format (named after the person who created the FASTA program and ditto format).
The format of the FASTA sequence is divided into three parts:
- After the carat sign ‘>’, SeqID is present, which is unique for each nucleotide or protein sequence and should not contain any spaces. The SeqID is limited to 25 characters or less. It can only include letters, digits, hyphens (-), underscores (_), periods (.), colons (:), asterisks (*), and number signs (#). The sequence identifier will be replaced with an accession number by the database staff when your submission is processed.
For instance, >SeqABCD
- The information about the source organism from which the sequence was obtained follows the SeqID. The name of the organism should be scientific.
For instance, >SeqABCD Rattus rattus
- The final optional component of the FASTA definition line is the sequence title, which contains a brief description of the sequence.
For instance, >SeqABCD Rattus rattus isolate New Zealand chromosome 9, Rrattus_CSIRO_v1, whole genome shotgun sequence
Let’s understand it with a real example.
>NC_000024.10:c2787682-2786855 Homo sapiens chromosome Y, GRCh38.p14 Primary Assembly
This is the FASTA format of the STY gene, here the FASTA starts with the “>” with a unique ID “NC_000024.10”, the gene is located between the nucleotides c2787682 to 2786855. It’s located on the human Y chromosome with a GRCh38.p14 primary assembly.
After that, the complete FASTA sequence is located for the SRY gene.
How to extract the FASTA sequence?
After having understood what exactly the FASTA sequence is and its format, let us now extract the FASTA sequence from the NCBI database following this step-by-step procedure.
Step 1: Open Google, and enter NCBI in the search box.
Step 2: Once you have clicked on NCBI, the window of the NCBI site opens.
Now, in the “search box” enter the name of the gene e.g. GAPDH whose information you wish to collect.
Besides the search box, there is a tab showing all databases, click on that tab and select “Gene” from the dropdown menu.
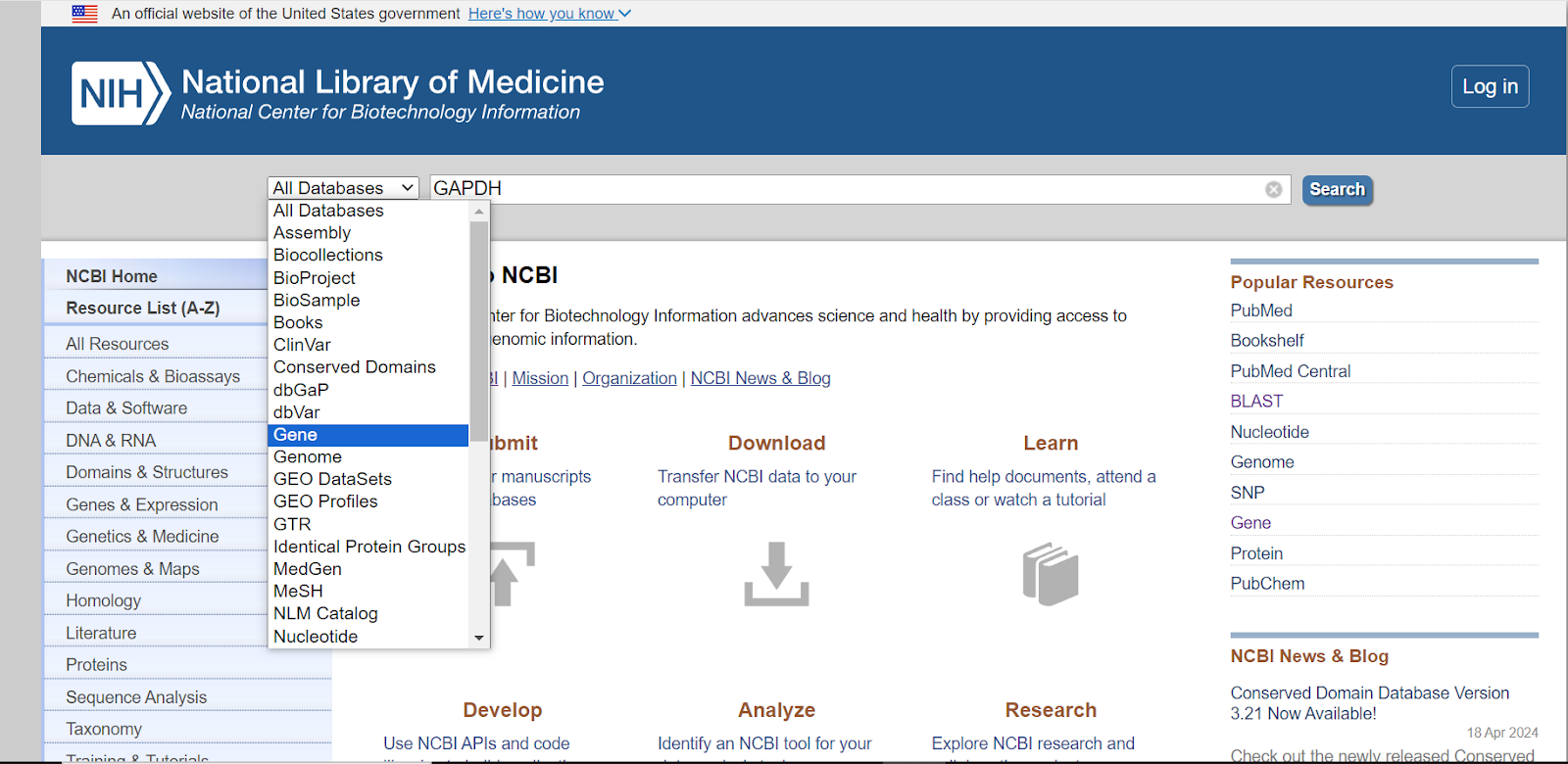
Step 3: Once you have selected Gene from the dropdown menu, click on the “search” button.
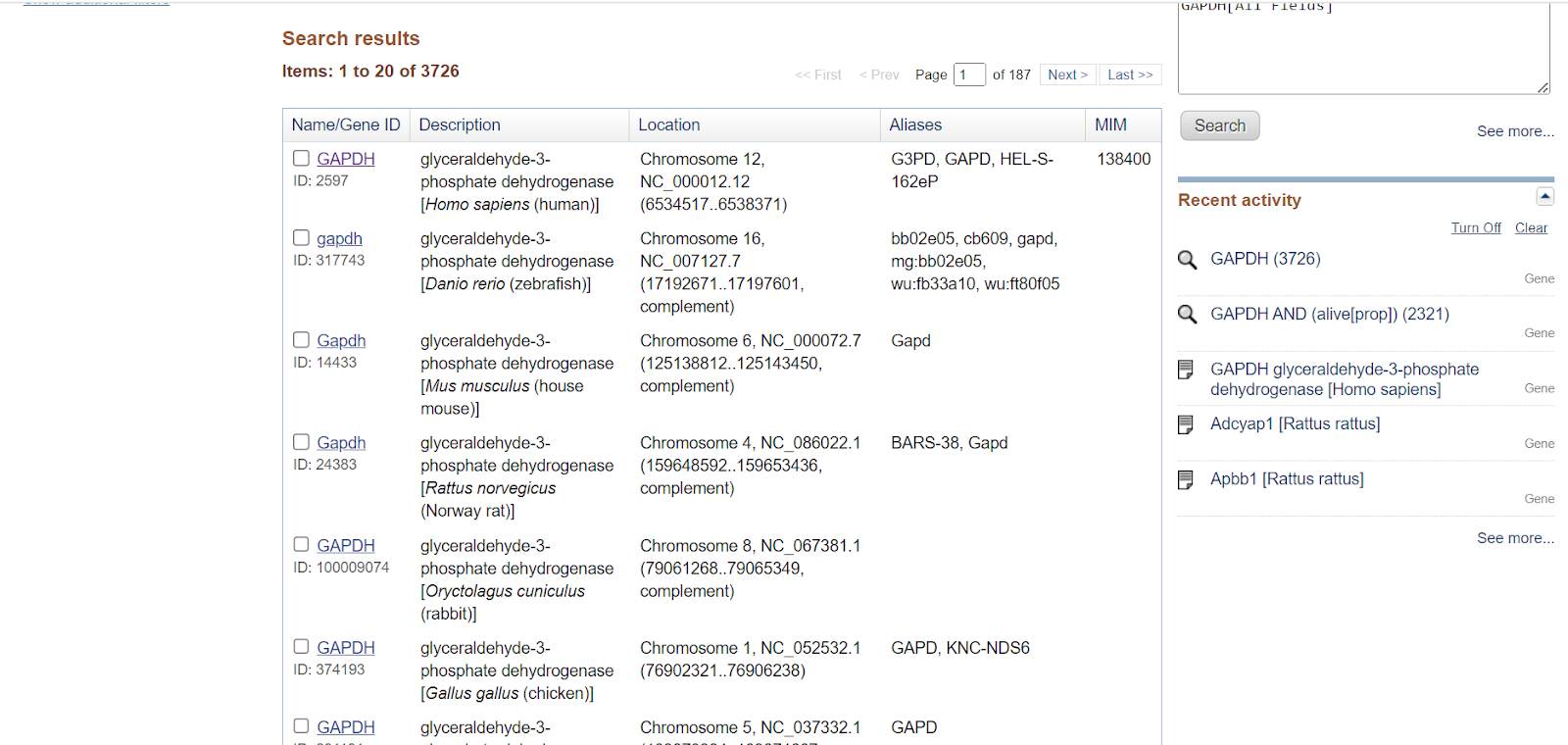
You will get a list of GAPDH genes present in various organisms. You can select a specific organism from the Top Organisms list present on the right-hand side of the page.
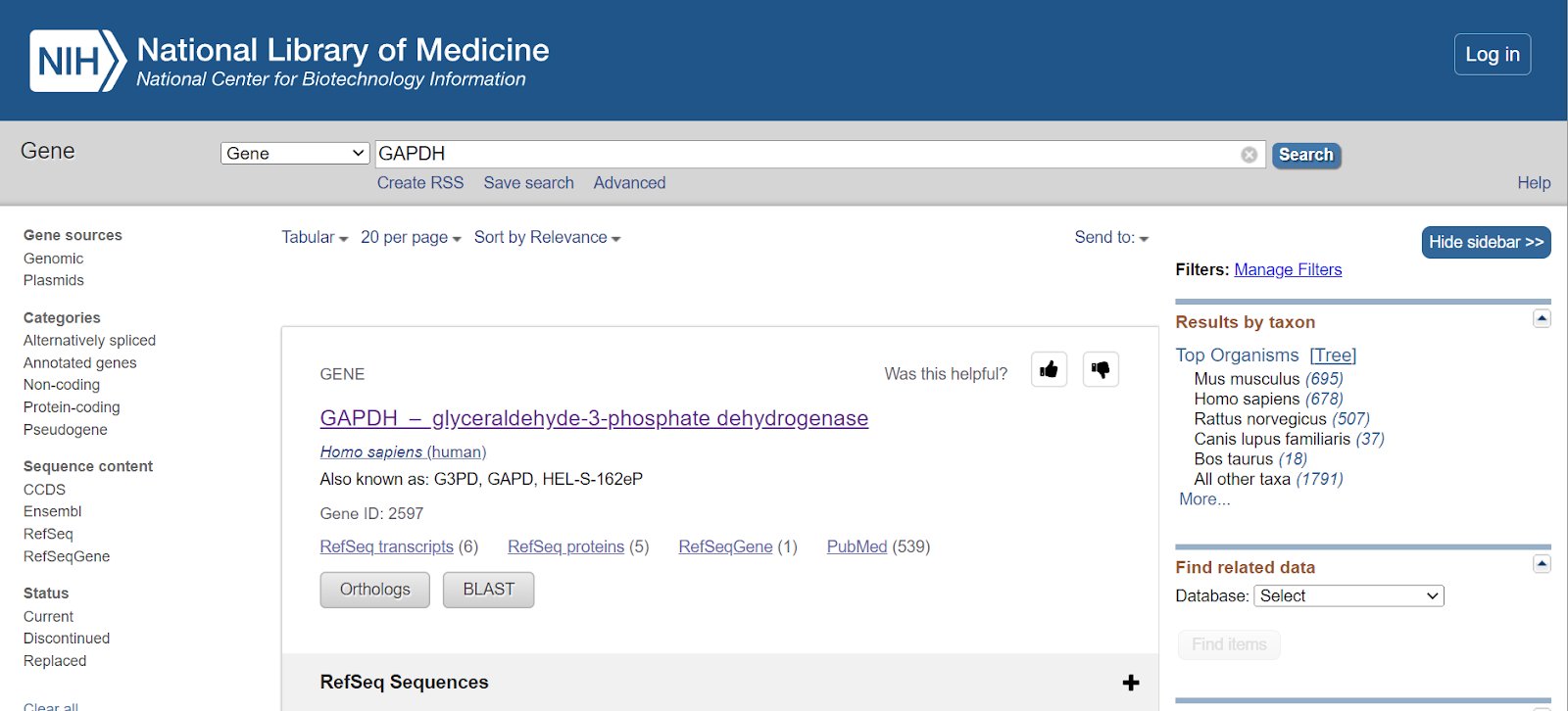
Step 4: Once you select the species from the top organisms, genes will be shown for only that particular species. E.g., here I have selected Homo sapiens, so all the genes related to GAPDH in Homo sapiens would only be displayed.
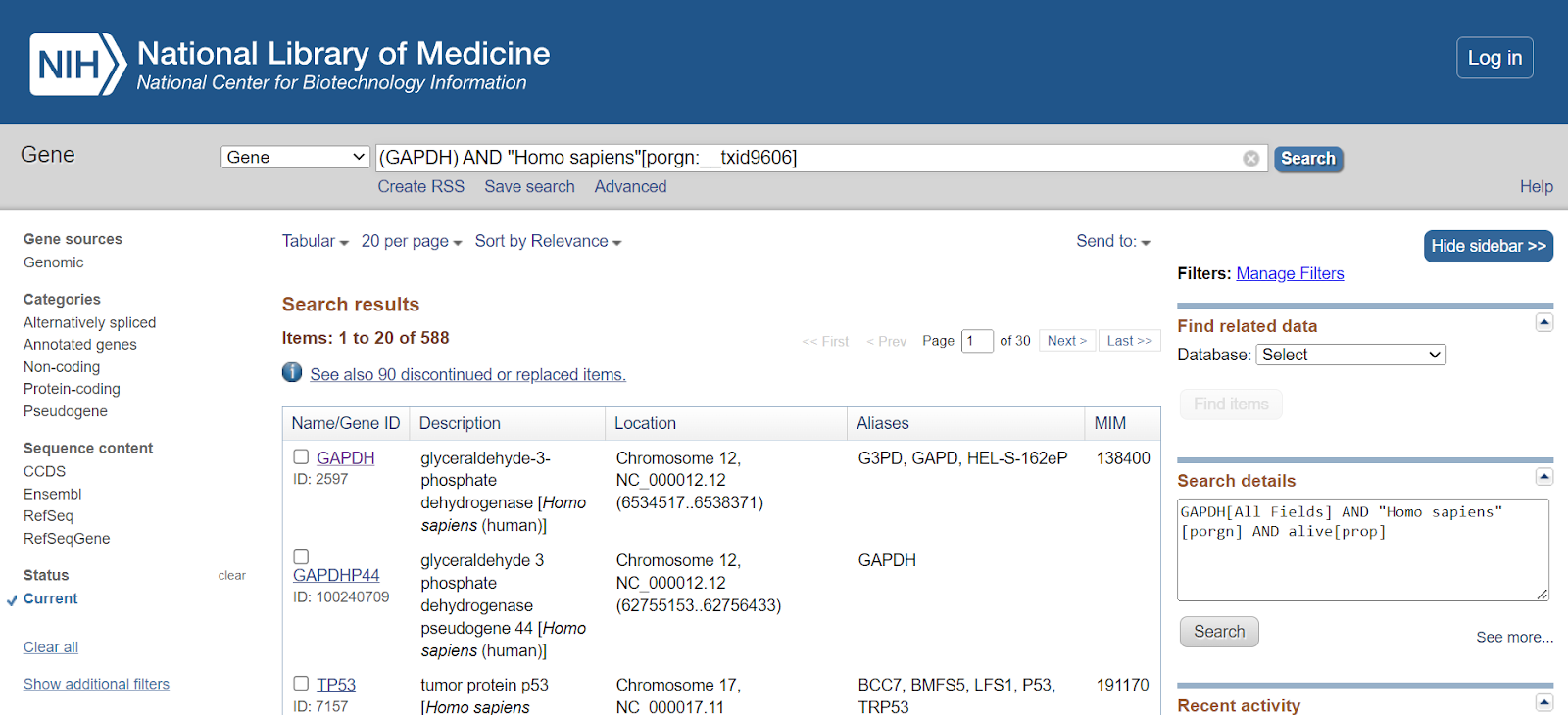
Step 5: Select the gene of your interest from the list.
Once done, the page containing gene information will be displayed. This will showcase the official gene symbol, the full name of the gene, gene type, organism, lineage, and a summary of the gene.
As you scroll down through the page, other information related to the gene can be obtained such as location and exon count.
The sequence of the gene can be retrieved in the form of a FASTA sequence. Click on the FASTA tab.
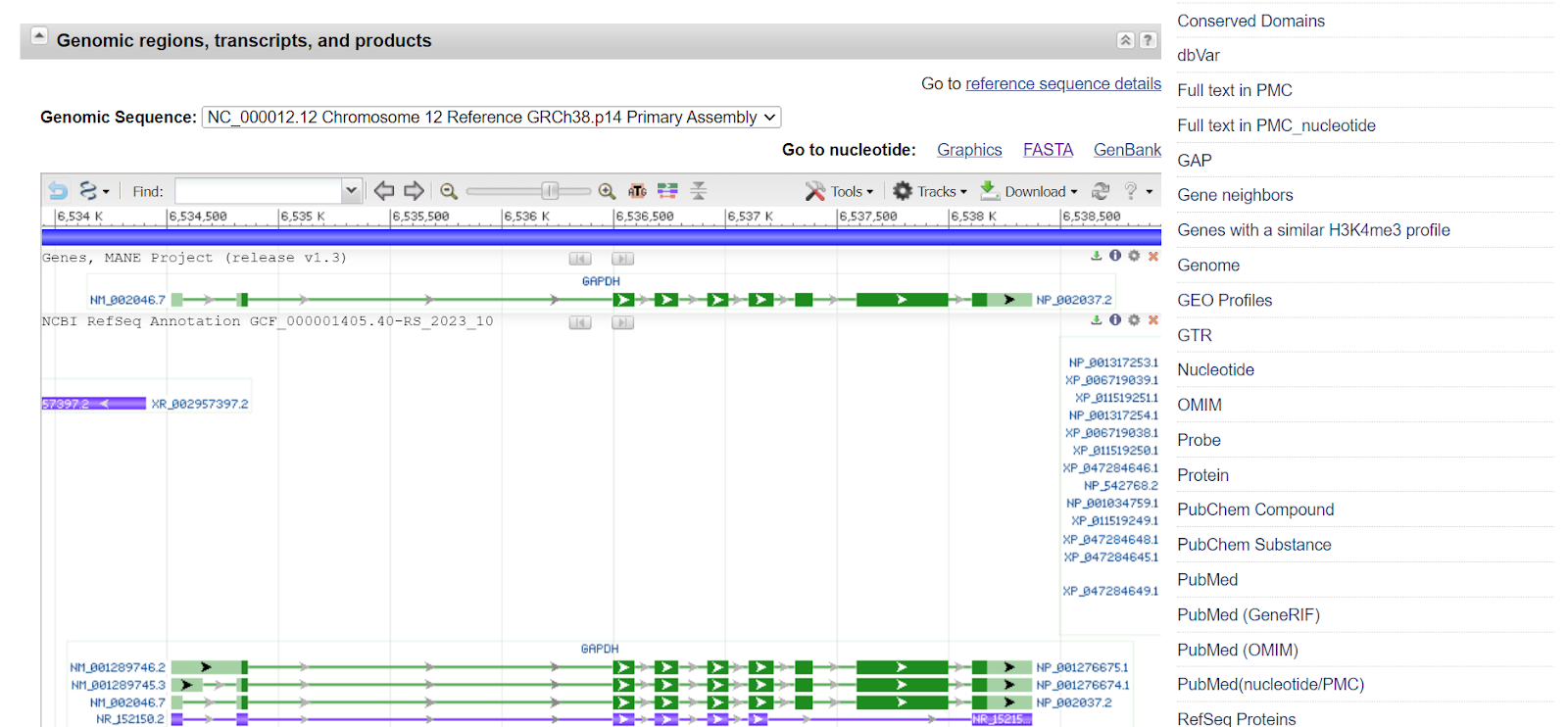
As you will select the FASTA tab, a new window will open showing the FASTA sequence.
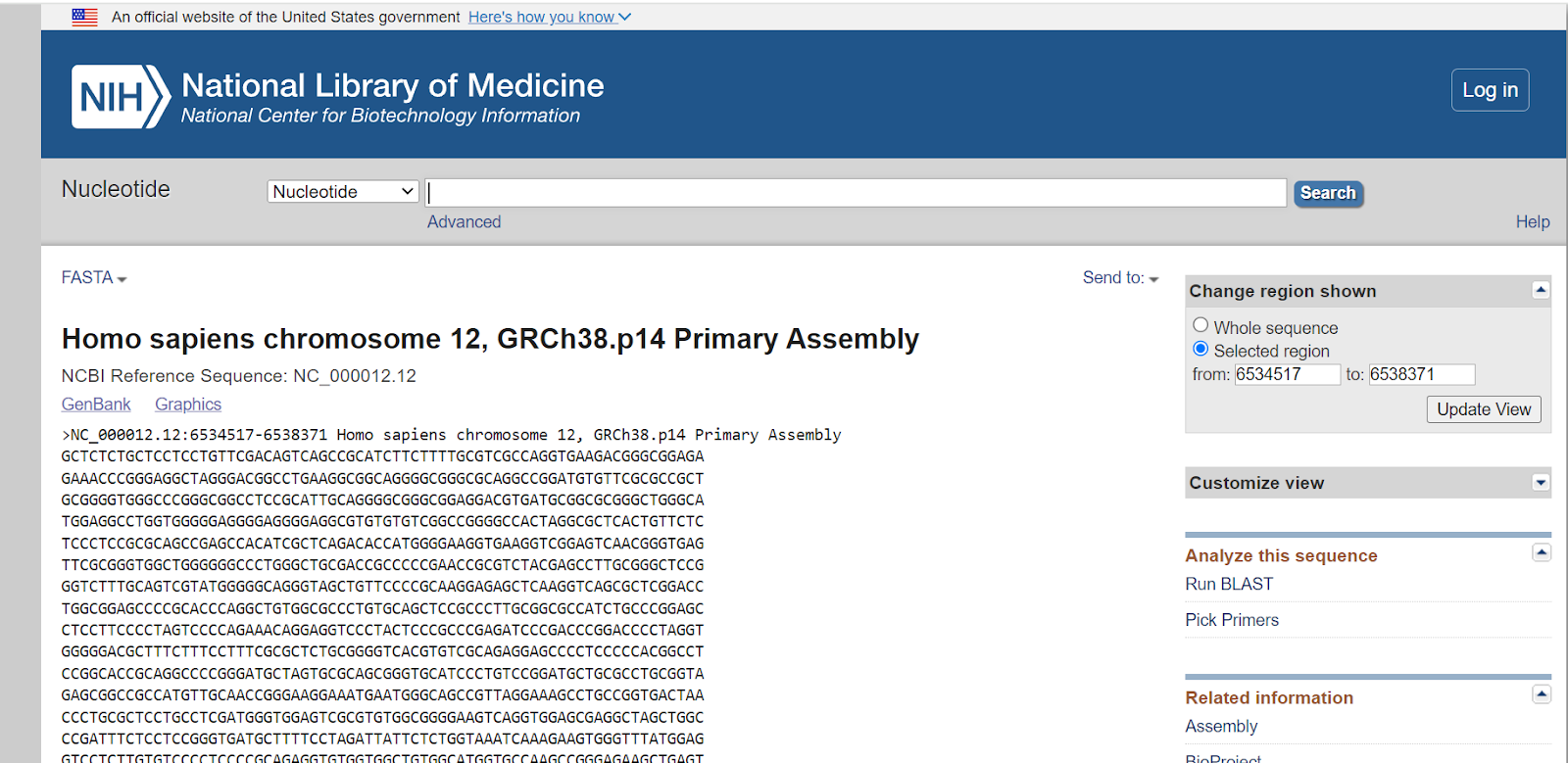
Here’s the FASTA sequence of the GAPDH gene.
How do you use the FASTA sequence?
Once the FASTA sequence is extracted from the NCBI database, it can be used in several ways. Let’s check them out one by one.
To design primers for PCR
Primers are short, single-stranded DNA sequences used in the polymerase chain reaction (PCR). PCR is used to amplify a specific gene of interest (GOI) using primers specific to the gene of interest.
Primers can be designed using various free software available online such as Primer 3, Primer-BLAST, Primer 3 Plus, PrimerQuest, and many more. All these programs require the sequence of the gene of interest to design primers specific to it. Therefore, the FASTA sequence of the GOI becomes useful here.
Related article: How to Design PCR Primers.
DNA sequencing
FASTA has a crucial role in DNA sequencing as well. Much like the PCR, here also, it is used to design the sequencing primers. In addition to that, the FASTA file works as a reference sequence to align sequencing reads.
Do you know?
The sequencing output obtained after the run is in the FASTA format, known as the FASTQ file. FASTQ contains the sequence data and the read quality data.
DNA microarray
In DNA microarray technology, the FASTA file is used to design various probes to immobilize the solid support. Probes are short DNA sequences complementary to the target DNA or gene and are labeled with a detectable tag.
Millions of probes are immobilized on the microarray chip. Using the FASTA file, these probes are designed.
Furthermore, it is also used as a quality purpose to validate the hybridization data.
Related article: 6 Types of Microarray-based Genetic Testing.
To perform Multiple Sequence Alignment (MSA)
Multiple Sequence Alignment (MSA) generally refers to the alignment of three or more biological sequences (protein or nucleic acid) of similar length. From the output, homology can be inferred, and the evolutionary relationships between the sequences can be studied. Various online free tools are available to carry out MSA, such as Clustal Omega, EMBOSS Cons, Kalign, MAFFT, MUSCLE, MView, T-Coffee, and WebPRANK.
Users are required to provide the FASTA sequence of the genes or proteins of all those organisms whose evolutionary relationships they wish to study. It has become quite useful in evolutionary studies.
To find the Open Reading Frames (ORFs)
The region of the nucleotide sequences from the start codon (ATG) to the stop codon is called the Open Reading frame (ORF). An ORF is a sequence of DNA that starts with the start codon “ATG” (not always) and ends with any of the three termination codons (TAA, TAG, or TGA).
Depending on the starting point, there are six possible ways (three on the forward strand and three on the complementary strand) of translating any nucleotide sequence into an amino acid sequence according to the genetic code. These are called reading frames. The ORFs are the regions that get translated into proteins with the help of ribosome machinery.
To find out the number of ORFs in a particular gene, an NCBI tool such as ORF finder is employed. ORF finder tool requires the user to enter the FASTA sequence of the gene of interest (GOI). Once entered, the tool provides a list of all ORFs present in that gene, along with the starting point, end point, frame, strand, and length.
Construct sequence logo
A sequence logo is a graphical display of a multiple sequence alignment consisting of color-coded stacks of letters representing nucleotides at successive positions. Sequence logos provide a richer and more precise description of sequence similarity than consensus sequences and can rapidly reveal significant features of the alignment that could otherwise be difficult to perceive.
The total height of a logo position depends on the degree of conservation in the corresponding multiple sequence alignment column. Very conserved alignment columns produce high logo positions. The height of each letter in the logo position is proportional to the observed frequency of the corresponding amino acid in the alignment column.
The letter of each stack is ordered from most frequent to least frequent so that it is possible to read the consensus sequence from the top of the stacks. The Web Logo tool is used to generate sequence logos. The user needs to provide multiple nucleotide FASTA sequences to generate it.
To locate restriction sites in DNA Sequences using the Restriction Mapper
Restriction Mapper sites for restriction enzymes, i.e., restriction endonucleases, in DNA sequences. Once the FASTA sequence of the GOI is entered, it gives a list of all restriction enzymes whose restriction sites are present within the GOI, along with information such as site length, site sequence, frequency, and cut positions.
It even provides a list of noncutters. Moreover, it even does virtual digestion. This information becomes very useful for designing experiments involving restriction enzymes.
Related article:
How To Choose A Restriction Enzyme?- Step-by-step Demonstration.
To perform BLAST
FASTA is used to perform Basic Local Alignment Search using the BLAST. Here it is used to align with various other sequences, genomes or databases. BLAST is somehow to similar to Primer3 software. The FASTA query can be directly pasted in the search box to perform the BLAST analysis.
Wrapping Up
In conclusion, FASTA is an important file format for various genetic experiments starting from the basics. The article covers the information on the FASTA sequence necessary for the readers to know.
I hope you enjoyed this article. Do share it and subscribe to our blog.