“Shotgun sequencing is a cutting-edge Next-Generation sequencing technology used in metagenomic analysis to sequence entire nucleic acid pool, allowing the identification and characterization of microbes present in environmental samples.”
Do you know?
Only 2% of bacteria can be cultured.
A Plethora of microbes is present in any environmental or biological samples. We can comprehensively understand any environment or ecosystem by metagenomic (taxonomic and functional) analysis. Taxonomic analysis avails us information on the identity of microbes while functional analysis avails us information on the role of each microbe in the ecosystem.
Metagenomic studies become powerful only after the recent advancements in the NGS platform. Shotgun sequencing, in particular, is the choice of technique for large-scale analysis in this field. It allows us to do diversity and functional investigations more accurately.
The simpler sequencing setup with steps– DNA isolation, library preparation and sequencing; and complex analysis processing with steps– assembly, binning and prediction collectively provide a snapshot of the entire microbial community.
Let’s see the role of the NGS platform and shotgun sequencing in metagenomic analysis and profound knowledge of its advantages, applications and limitations.
Recommended read: What is Metagenomics?
Stay tuned.
Key Topics:
What is shotgun sequencing?
Shotgun sequencing is a state-of-art, high-throughput and next-generation technique that can sequence the entire genome. Hence, utilized extensively for human genome sequencing. However, here, by considering the entire genetic content of the environmental/biological sample as a “genome” every nucleotide is sequenced.
The process is though a bit different than conventional genome sequencing but provides all the advantages of next-generation sequencing for large-scale microbial analysis. Like the speed, precision, sequencing depth and higher coverage rate.
Due to such important and lucrative benefits, shotgun sequencing is now widely used in metagenomic analysis. The most important reason why shotgun sequencing is the ultimate “choice of technology” is because of its ability to find and sequence low abundance sequences from the sample.
Now let’s understand the process and steps. Keep in mind, our objective is to describe the technique in the context of metagenomics only, we would explain the original technique in some other articles.
Shotgun sequencing steps
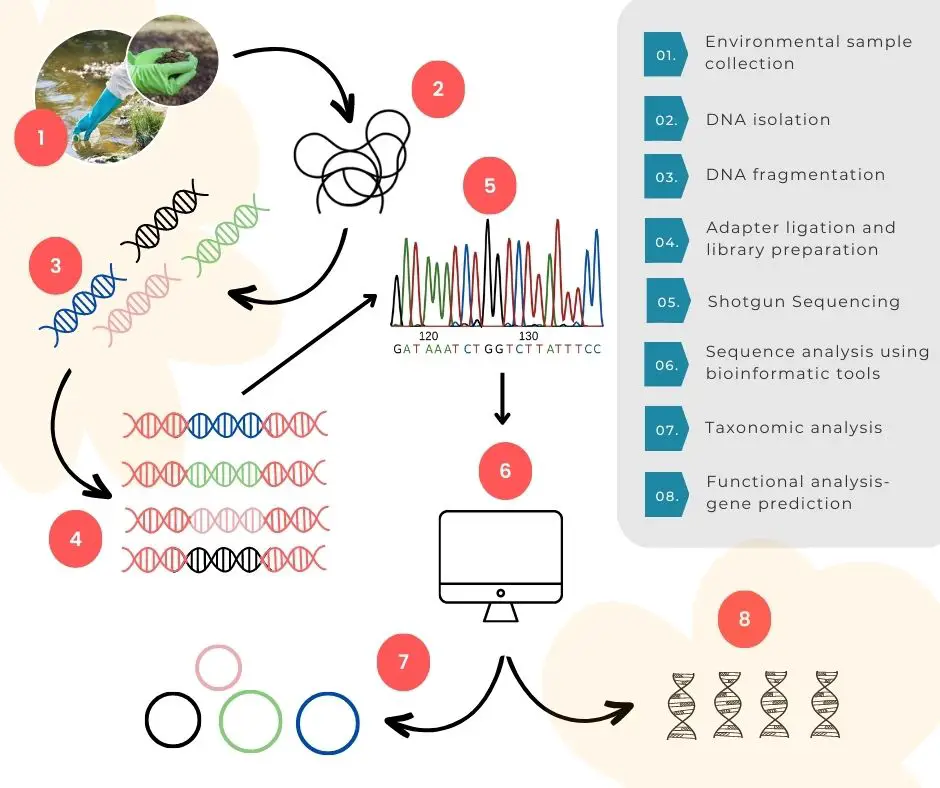
Sampling and pre-processing
Any environmental or biological sample is processed for DNA extraction, quality and quantitative analysis and sent for fragmentation. It’s often important to choose the correct DNA extraction scheme for metagenomic analysis, unlike conventional DNA extraction.
Any metagenomic sample is rich with enormous microbes having an unknown identity so the given technique can isolate DNA from “some” but not from “all” microbes from the sample which is likely to occur. Therefore, a greater and most diverse part of the microbial community may remain unexplored.
Manual chemical methods definitively can’t be the choice but even the highly advantageous spin-column DNA extraction techniques can’t do the job. It’s important to understand here that the main objective for metagenomic DNA extraction is not only to obtain a higher yield but also to isolate DNA from every possible microbe present in the sample.
Bead-beating and magnetic bead-based extraction are great options for effective metagenomic DNA isolation. However, it may also cause DNA loss. Go through this checklist before selecting a nucleic acid isolation technique for any metagenomic sample.
- Should yield high DNA.
- Should yield highly pure DNA.
- Capable enough to isolate DNA from every microbe present in the sample.
- Should avoid any contamination.
- Can’t isolate host DNA.
Library preparation
The main advantage of next-generation sequencing is precision which is acquired by reading smaller fragments one after another. DNA is fragmented into smaller-sized fragments until overlapping fragments are generated.
Overlapping fragments help in assembling sequences. The more the number of overlapping fragments, the more precise the assembling process is. One subsidiary step during this process is enrichment.
The library of fragments is enriched– the desired no. of fragments is generated by PCR amplification. Random and known hexamer primers are annealed with the sequence and allowed for amplification. However, PCR bias is a common problem here.
In the last step, known adapter sequences are ligated with the target sequences for sequencing. Adaptors are known sequences for the machine.
Technical consideration:
PCR amplification-free library preparation is highly recommended for preparing metagenomic DNA libraries as amplification sometimes over-amplifies some fragments which creates results bias. Kits are now available for such preparation.
Sequencing
Now sequencing occurs in a massively parallel fashion. The sequence settled on the solid surface and each sequence in the sample is identified and sequenced. After completion of the sequencing process, the data is sent for assembly.
Read length is an empirical technical parameter taken into consideration for sequencing the metagenomic sample. Long read lengths have a higher error rate, low-sequencing depth and high cost. Short read length provides considerably more valuable results at a low cost.
To my knowledge, long-read sequencing is ideally applicable to investigate “some” and “known” microbes from the sample. For random analysis or when organisms are unknown to us, a short read is preferred.
Related article: DNA sequencing: History, Steps, Process, Methods and Applications.
Assembly
For assembling the entire sequence, a computer program takes the help of overlapping sequences. The program finds the many overlapping fragments and arranges the sequence accordingly. The assembly read depth should be at least 15X. Meaning, each nucleotide should be present/sequenced at least 15 times.
The higher the read depth, the higher the accuracy and precision. Once the entire nucleotide bunch is assembled, it is processed for taxonomic and functional analysis.
Analysis
Bioinformaticians analyze the sequence for phylogeny, diversity and functional analysis. Binning and annotation are common in metagenomic analysis which finds genes and their functions. Many analysis tools are now available and provide diverse information regarding the microbial population present in the sample.
Advantages
- The present NGS platform is more advantageous for environmental DNA sample metagenomic analysis. As it reads every bit of nucleotides present in the sample and allows comprehensive analysis of various microorganisms.
- It also provides de novo and assembly-based sequencing analysis for taxonomic and functional evaluation of data. Meaning, the data can be compared with the pre-existing sequence information for identification characterization.
- Assembly by generating counting allows functional analysis by determining the known and unknown sequences present in the sample.
- Overlapping fragments allow researchers to accurately and precisely locate each sequence with the correct one. This feature gives shotgun sequencing immense accuracy and precision.
- Shorted read-length sequencing is more feasible as the machine can read each nucleotide correctly.
- Extensive amplification is not required which is yet another advantage for achieving excellent sequencing depth (for each sequence including non-abundant sequences too), and decreasing the cost and time for sequencing.
- The present NGS workflow can even find low-abundance sequences i.e. low abundant microorganisms from the sample and construct their potential functional profile.
- It’s even superior to the conventional 16s rRNA gene sequencing as not only some part but the entire gene is sequenced and allows classification at an even smaller (species) level. Thus it can detect bacteria, viruses, fungi, archaea and even eukaryotes present in the sample.
- In addition, it is speedy and requires less starting material and pre-preparation compared to other platforms.
- The present tool adds more value in revealing microbial dark matter. Microbial dark matter is referred to as the pool or sub-community of microbes which is still unknown to us. However, due to the unavailability of the reference genome, more information can’t be collected.
- The present technology is so sensitive that it can even perform sequencing using a small amount of starting material. The present advantage becomes so important when we analyze crucial, rare and low-abundant samples.
- It can work for low biomass metagenomic samples and thus eliminate the need for traditional culture or cultivation. Additionally, microbes that can’t be cultured are sequenced using shotgun sequencing.
Disadvantages
- Shotgun sequencing is a very costly method. Whereas the conventional 16s rRNA gene sequencing technique cost around $50 to $80, shotgun (NGS) cost between $200 to $500 which depends on the read depth required.
- It also requires high-end and superior computational facilities, additional software and skilled manpower.
- The data obtained is also huge and very complex. High-end and powerful computers and computational tools as well as skilled personnel are required to process and analyze the data.
- One of the major limitations of shotgun sequencing is that repetitive sequences can’t be assembled correctly as the repeat units interfere with overlapping.
- It can’t eliminate the host DNA sequences from the reads. If the sample is derived from a human, animal or plant host microbiome, the host DNA is also extracted and sequenced which makes the overall interpretation and assembly process inaccurate.
- No sufficient shotgun and NGS data are still available to compare and draw conclusions with the available samples.
Challenges and Future Prospects
Most of the time, the major problem every geneticist faces is the lack of reference genome or sequence as every microbial community and/or metagenomic sample is highly diverse. In recent times, however, there are so many metagenomic databases available.
Metagenomic assembly is a difficult process as it’s a mixture of low and high-abundance genomes. Also, it contains various strains of bacteria and strains with single or a few nucleotide changes. Such problems can be overcome by increasing sequencing depth.
However, the absence of a fragment of any gene, operon or gaps in the sequence is still difficult to determine and fill.
In trends, shotgun metagenomic sequencing is yet not extensively applicable and ready to penetrate into the medical field and clinical diagnosis. Here are some of the reasons for that.
The high sensitivity of the present technique is also a limitation of it. It can even identify and predict a single microbe present in a clinical sample, where any other available techniques provide negative results for the same sample.
The problem is that it can even sequence host/human DNA present in the clinical sample which extremely restricts the use of metagenomic NGS in clinical setups. Available tools, right now, have limited capacity to post-process contaminated sequences. In addition, standards or controls are yet not available for evaluation, which again limits its use in clinical and medical testing.
Another problem also raises the question of whether the sequence present in the sample has any direct relevance to the patient’s disease condition or not. Because the sample may be contaminated during collection, transportation or processing.
Also, any novel strain or infection, if found in the sample, is directly linked with the existing health condition or not. Because we right now have fewer data regarding metagenomics. Nonetheless, despite these challenges, NGS-based shotgun metagenomic sequencing has pivotal significance in other fields.
Right now the technology is yet not fully ready to use in medical testing and diagnosis.
Do you know?
There are NGS platforms like HiSeq, NovaSeq or NextSeq that can produce 100 GB to 1.5 TB data per run. Now imagine how powerful computer we require!
Wrapping up:
Metagenomics is growing at a rapid pace. More and more research is available every day, databases are being prepared and tools and pipelines are standardized. So in near future, we can use such a powerful tool in the medical field too.
The use of shotgun sequencing and the NGS platform indeed revolutionized the entire sub-discipline and, newer methods and dedicated platforms are, further, under construction. Notwithstanding, shotgun metagenomic sequencing proved as a powerful tool in ecological, environmental and agricultural research.
I hope you like this article. If you want to strengthen your knowledge on the present topic, read our previous article. Share this article.

