“A rapid colony screening method, the colony PCR validates the target DNA or gene construct present in a bacteria that has grown on selective media.“
As a result, researchers determine the presence or absence of DNA inserts in a plasmid directly from bacterial colonies.
PCR- The polymerase chain reaction has many good variants, (unlike the loci variants from Marvel!). Each variant has been developed to address a specific problem. For example, multiplex PCR saves time and cost.
Besides conventional PCR; Multiplex PCR, qPCR, RT-PCR, Allele-specific PCR, Immuno PCR, methylation PCR and long-Rang PCR are a few common variants. The colony PCR technique is one among the lists that has specific importance in molecular or gene cloning experiments.
PCR has many diverse applications ranging from amplifying a gene to validating an insert DNA. In colony PCR using the insert-specific primer set, scientists can validate a new gene or insert DNA in a plasmid.
The novel method tells us whether the gene of interest is transformed into a bacterial plasmid or not. Although, traditionally scientists have to heavily rely on transformation followed by restriction digestion.
The transformation experiment is a time-consuming process, costly and less accurate. Restriction digestion needs a special set up too. On the other hand, colony PCR assay does do the same work in less time!
The process is a bit different and quite difficult, but I will explain things in layman. This article will explain the technique colony PCR, its principle, process, protocol, applications, and limitations.
Again, this article will add more value to your PCR knowledge and helps you to achieve more success in PCR technique,
Stay tuned,
Key Topics:
What is Colony PCR?
It’s a modification of a conventional PCR including denaturation, annealing and extension steps and amplifies DNA by the cyclic process. Colony PCR has been utilized in gene transfer, gene cloning and genetic engineering experiments to validate the insert DNA.
It’s high throughput, rapid and cost-effective technique that amplifies only if the gene of interest/inserts DNA/foreign gene is present.
Meaning, it works only when the transformation is successful. The main advantage is using bacterial colonies directly as template DNA.
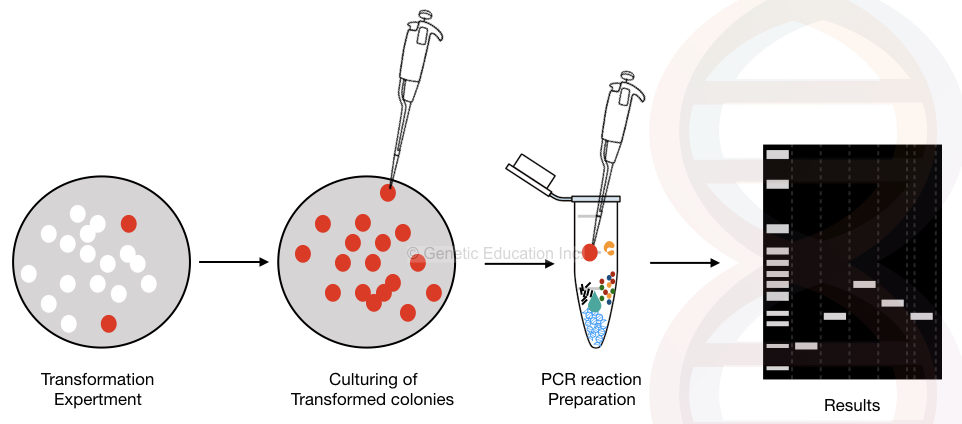
We can use bacterial colonies directly as template DNA. Cyclic-enzymatic-temperature-dependent reaction only amplifies successfully transformed plasmids. The rough draft of the process is explained in the figure below,
Before going ahead in the article, we first have to understand the concept of plasmid DNA. Until you don’t know what a plasmid is! You don’t understand the topic.
What is a plasmid?
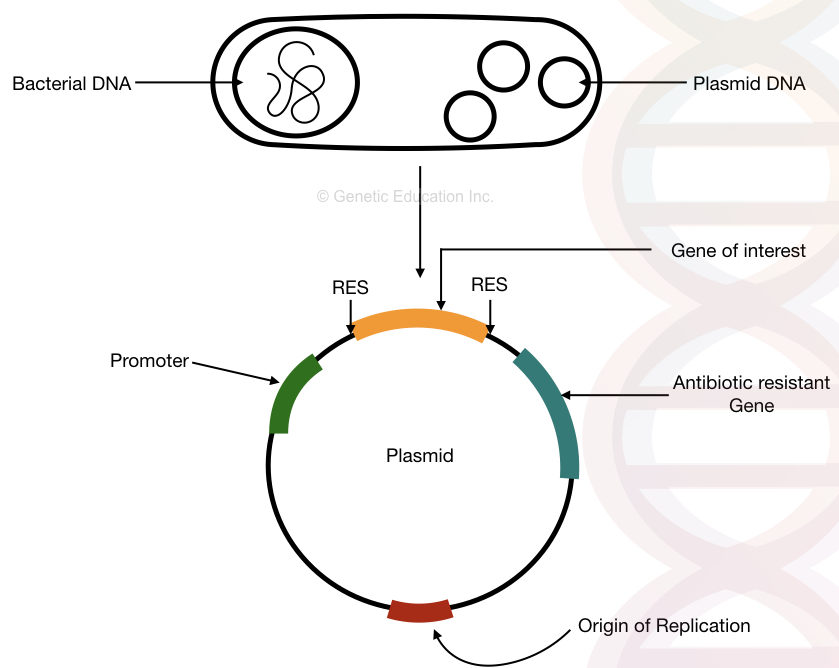
“A plasmid is the bacterial circular DNA which replicates independently from the bacterial chromosome and is used in the gene manipulation and gene transfer.”
Plasmids present freely in bacteria and have a meaningful role in genetic engineering. Scientists use the plasmid as a vehicle to transfer a gene or interest, popularly known as the insert DNA.
The plasmid is a small, circular dsDNA of bacteria. Physical, chemical and artificial techniques transfer a GOI in a plasmid.
When replicated independently, it forms many copies of our genes of interest. Because bacteria replicate faster, we get a huge number of copies.
Plasmids have genes that help bacteria to survive in harsh and unfavorable conditions. Several other prokaryotes also possess plasmid DNA besides bacteria.
It works as a gene carrier to transfer a gene. Several common types of plasmid are F-plasmid, Col-plasmid, degradative-plasmid and resistance plasmid. The figure below, roughly explains the plasmid.
Traditionally, validating insert DNA relies on the cell culture technique in which scientists culture bacteria on a plate for 3 to 4 days. As we said, the cell culture technique has serious limitations.
In another method, an additional step of restriction digestion has been added. In RE, restriction endonuclease cuts DNA at a specific location using which we can validate the insert. Both techniques aren’t suitable, accurate and reliable.
A PCR can validate or assess a GOI in a single PCR reaction, using direct cultured cells, that’s what the colony PCR is!
The technique colony PCR is accurate, fast, and sensitive enough to detect GOI. It can also quantify the DNA as well. Now get back to the principle colony PCR,
Principle of Colony PCR:
After transformation, bacterial colonies have been picked and added to the PCR reaction. The reaction already has mastermix- a cocktail of dNTPs, PCR reaction buffer, Taq DNA polymerase, primer set and nuclease-free water.
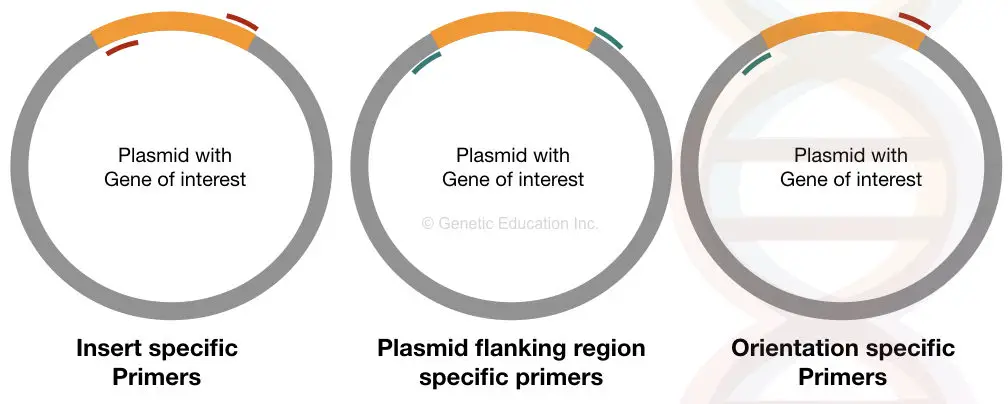
Two sets of primers amplify two different plasmid DNA, the insert-specific primers amplify the gene of interest whereas the vector-specific primer amplifies the flanking region along with the insert.
Both primer sets amplify DNA in the same direction but have a different purpose. Sequence primers note the presence or absence of the insert while Flanking primers help to determine the size of insert DNA on a plasmid and that’s why important.
After completion of the PCR, agarose gel electrophoresis helps to evaluate the result. DNA sequencing identifies the inset correctly.
This is the basic colony PCR protocol. Although, we can modify the protocol as per assay requirements.
Colony PCR procedure:
We can divide the process into a few steps such as transformation, selection and designing primers, reaction preparation, PCR amplification and results evaluation.
After completion of gene transfer experiments, colonies can directly be used as a template DNA, so we don’t require any DNA extraction.
You may wonder why we don’t need to extract DNA here? How can plasmid DNA be amplified directly? The answer is simple.
The bacteria has a smooth and soft cell membrane that degrades only by heating. Centrifuging a sample or heating can lyse the cell membrane and release the plasmid.
In addition, we don’t need the bacterial chromosomal DNA too, that is why only including a single heating step in PCR works fine for colony PCR.
Furthermore, additional purification steps aren’t required as we only need plasmid DNA. We can avoid alcohol purification and can save time and cost.
Primer designing has great importance in the process, of what information we should need, depending upon that, primers are developed. For example,
- Information about the presence or absence of the insert only.
- Information about the size of the insert.
- Information about the orientation of the insert.
Based on these three types of information, different primer sets can researchers use are; insert specific primers, plasmid flanking region-specific primers and orientation-specific primers.
The graphical representation of how all three work, explained here in the figure below,
Let us understand a bit more about each type of primer.
The insert-specific primers have been designed specifically to amplify only the region comprising the insert DNA or GOI. It binds on both sides of the insert DNA and starts amplification inward.
If colonies contain a GOI, it binds and amplifies, otherwise, it can’t amplify when it finds nothing. This primer typically provides information regarding whether the ‘insert’ is present or not.
However, it can’t tell you if it is there in the plasmid or not. It also can’t show the size of the ‘insert’. It simply gives an idea regarding the presence or absence of the GOI or inserts.
It can’t even say anything about the orientation of the insert. Notedly, no amplification is shown in the negative control.
Orientation-specific primers have a unique function and so have a decent setup. Here, one primer binds inside of ‘insert’ whereas the other primer binds outside, on plasmid DNA.
The aim of doing this is to extract information if the insert is in the correct orientation or not! The orientation has great value in gene transfer experiments as the wrong orientation can’t give us the desirable output.
See the third section of the above figure carefully. Usually, it isn’t a different set of primers. One primer from the sequence-specific set and another primer from the flanking-region-specific primer can be utilized to do so.
The objective of making an orientation-specific primer is to check the orientation of the insert DNA, and whether our GOI is ligated correctly or not.
If anything goes wrong, orientation primers can’t amplify the plasmid, we can know that the plasmid isn’t ligated corrected. On the downside, it even can’t predict the size of the insert fragment.
Note that in the negative control, we can’t amplify.
The plasmid-specific primers are as important as the two explained here. Here the primers are designed in such a way that amplifies the flanking regions of the insert.
It typically gives an idea about the size of what we have inserted. The positive reaction shows a larger DNA band than the negative reaction.
To correctly evaluate the gene transfer experiment and assess the insert, all three primer sets are used in the experiment.
The primer should follow all the criteria required for a typical primer set. It should have low GC content, up to 20 bp length, low hairpin structure, and non-specific binding.
Summary:
| Primer set | Positive reaction | Negative reaction | Application |
| Insert-specific primers | Amplifies the insert DNA | No amplification | To check the presence or absence of insert. |
| Orientation specific primers | Amplifies insert and flanking DNA | No amplification | To check the orientation of the insert |
| Flanking region-specific primers | Amplifies the region flanking the insert. Larger amplification. | Smaller DNA amplification product. | To find out the size of the insert. |
Read this article to learn more regarding the present topic: PCR primer design guidelines.
After completing the primer design, a PCR reaction is performed.
The PCR reaction should have Taq DNA polymerase, dNTP mix, reaction buffer, nuclease-free water and forward & reverse primers.
After preparing the reaction, tubes are placed in the PCR machines to amplify. 30 to 40 cycles are enough to get good amplification.
After completion of the reaction, the results are run on the agarose gel electrophoresis. Usually, 2% gel is preferred. Following purification, tubes are sent for DNA sequencing. Sequencing helps to understand the sequence of insert as well as flanking regions.
Colony PCR protocol:
Pick several bacterial colonies using a sterile picker and transfer them into a PCR tube.
Add a TE buffer to it and heat the sample in a boiling water bath at 65C for 20 minutes. Note that you can also use nuclease-free water in place of TE buffer.
Gently vortex it.
Centrifuge the sample at high speed for 2 minutes.
Transfer the supernatant to another tube and use it as a template DNA.
A 20μL sample is added to the reaction.
Additional information:
Why supernatant and not pellet?
DNA is a biomolecule of life. The plasmid DNA is even smaller than the bacterial nuclear DNA. It contains only several genes of up to 1000bp to 20,000bp.
Hence by only centrifuging it, the lighter plasmid DNA comes out from the cell and settles in the supernatant while the pellet contains proteins and nuclear DNA remains on the bottom of the tube.
Prepare a PCR reaction as given into the table,
| Component | Concentration | Quantity |
| Master mix (Special for the Colony PCR) | 1X | 12µL |
| PCR reaction buffer With 2mM MgCl2* | 1X | 5µL |
| Forward primer | 10pM | 1µL |
| Reverse primer | 10pM | 1µL |
| Supernatant | 3µL | |
| Water | 3µL | |
| Total | ————————– | 25µL |
Add 12µL 1X mastermix or dNTPs mix.
Add 5 µL 1XPCR reaction buffer. Make sure to use the colony PCR-specific reaction buffer (available commercially). It must contain MgCl2.
Add 10 pM, 1µL of each primer to the reaction.
Add the supernatant (we have processed above) up to 3µL.
Add nuclease-free water as required to make the final reaction 25µL.
Put all the tubes in the PCR machine and set up the reaction as given in the table below.
| PCR Steps | Initial Denaturation | Denaturation | Annealing | Extension | Final extension |
| Temperature | 95 ̊C | 95 ̊C | 55-65 ̊C | 72 ̊C | 72 ̊C |
| Time | 3min | 10 sec | 45 sec | 50 sec | 5 min |
| ——————– | ——————- | 25 cycles | ————— | ——————— |
Advantages of colony PCR:
The technique is rapid and cost-effective.
It has more accuracy and specificity.
It’s a simple setup, just like the conventional PCR. DNA extraction and plasmid purification aren’t required.
Tedious, time-consuming and costlier techniques like restriction digestion can be avoided. Only a simple colony PCR can work finely and give the results that we want.
As we said, it’s a time-saving process, that completes within 90 minutes.
Disadvantages of colony PCR:
Undoubtedly, the colony PCR has dozens of advantages, however, it has a few downsides too. Here are some,
Any mutation or SNP within the ‘insert’ can’t be encountered.
It can’t give us sequence information, we need to rely on sequencing.
The chances of false-positive results are very high.
Applications of colony PCR:
Researchers use colony PCR in gene transfer, gene therapy, gene cloning, genetic alteration and CRISPR-CAS9-like experiments.
The main application of colony PCR is to evaluate the correct ligation process in bacteria and yeast.
Colony PCR optimization:
Designing a primer, preparing a reaction and doing amplification isn’t enough to get successful results in colony PCR. We need some extra optimization to achieve the goal.
Here are some of the tips you can use, or point you can take care of.
Using a few colonies, too much starting material increases the chance of non-specific amplification.
Use controls such as positive and negative controls to monitor the results. Controls are important to evaluate results.
Use flanking region primer set as a positive control, even if it lacks insert DNA, you can see a band of flaking primer amplification, meaning the reaction is well prepared.
As a negative control using the untransformed colonies, only flanking primer sets can amplify it, but not the insert primers.
In the gene transfer experiment select shorter DNA sequences as GOI, longer sequences elevate chances of non-specific bindings. Also, undesirable GOI fails reaction.
Use a shorter PCR program. Too many cycles do nothing as there will be nothing to amplify while fewer cycles abort amplification prematurely.
Run the reaction on 2% agarose gel for half an hour. You may get the banding pattern shown in the figure below,
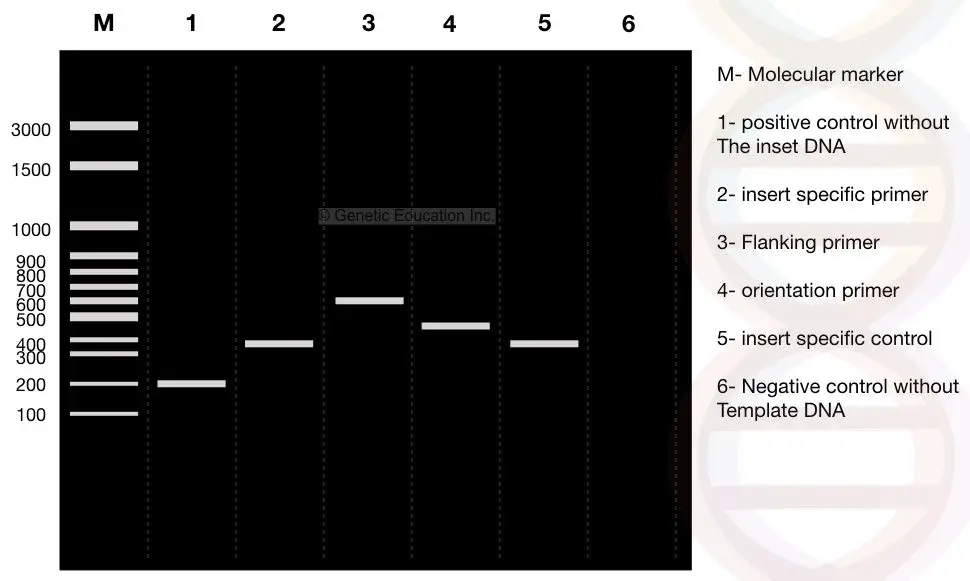
Colony PCR result analysis:
See the image carefully, I have prepared a conceptual gel image showing colony PCR results. I will explain the results of each lane separately.
The first lane is our DNA ladder of 3000bp and denoted as ‘M’. We use the DNA ladder as a reference to evaluate results. Read this article to learn more: What is the DNA ladder?
Assume that our insert is 400bp long. Lane 2 is our ‘insert’ amplified by the insert-specific primer. It’s 400 bp.
Lane 3 comprises amplicons of 600bp fragments, meaning it’s a fragment obtained by using the flanking region primers.
Lane 4 shows the results of orientation-specific primers. Note that the band size of orientation-specific primers is between the insert-specific and flanking-specific primers.
Lane 5 is the positive control for the insert-specific primer and is the 400bp fragment.
Lane 6 is the negative control for orientation-specific primers.
Lane 1 is the positive control for the flanking region-specific primers which is a 200bp fragment.
Wrapping up:
Colony PCR needs the direct use of colonies for amplification which is an accurate and faster method compared to previously available options. However, we need sequencing to understand and identify the insert.
The use of different primer sets makes it more tedious for routine use. In addition, interpreting results needs an expert touch.
I hope you like this article. If it helped you please ‘bookmark’ it on your browser.
Sources:
Jamal MAHM, Sharma SP, Chung HJ, Kim HJ, Hong ST, Lee S. Ultra-High Efficient Colony PCR for High Throughput Screening of Bacterial Genes. Indian J Microbiol 2017;57(3):365–369.