“Steps in preparing DNA for NGS and any other sequencing platforms are DNA isolation, DNA yield and quality check, library preparation, amplification and sequencing. The success of entire experiments highly relies on how each of these steps is performed.”
High-throughput sequencing has been increasingly utilized to sequence the genomes of many organisms with unprecedented accuracy. It reveals the genetic makeup of the sample by reading each nucleotide present. Give us an idea about sequence alterations, resultantly.
One crucial factor having a direct effect on the sequencing efficiency is the DNA sample preparation. Any sequencing method is a highly sensitive genetic technique and therefore is more error-prone. So every step must be performed carefully and accurately to obtain high-quality data/reads.
In this article, I will explain how to prepare a DNA sample for sequencing or NGS to succeed in the experiment and get high-quality reads.
Key Topics:
How to prepare a DNA sample for sequencing?
Keep in mind that we are discussing the scheme in general. All the recent day-high throughput and Next-generation sequencing would follow nearly the same scheme. However, minor to some crucial variations can remain, which vary depending upon the platform.
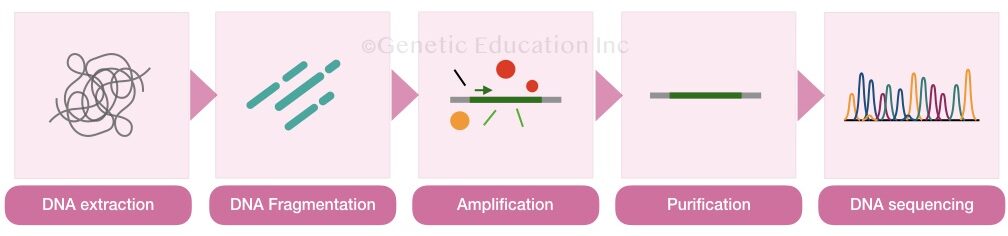
Sample preparation is explained here in steps.
DNA isolation:
Not only sequencing but any genetic experiment initiates with DNA isolation. DNA can be obtained from any biological samples– like animal tissues, plants, bacteria, viruses or even environmental samples.
Various techniques are available for the isolation of DNA, although chemical-based extraction techniques like PCI, proteinase K or CTAB are popular among the list. Notedly, the DNA isolation technique can be selected depending on the type of sample.
However, in general, to collect high-quality DNA for sequencing, manufacturers recommend using ready-to-use kits based on spin-column or magnetic bead-based technology. If you would like to know the differences between each technique, read this article: Comparison between manual: Spin-column and magnetic Automated DNA isolation.
Regardless of the isolation method chosen, the goal here is to obtain high-quality, high-yield and intact DNA. The isolated DNA should be free from any contamination. Contaminants interfere with the sequencing reaction.
Quality and quantity assessment:
High-yield DNA with a purity of nearly 1.80 at a 260/280 ratio is the ideal DNA for sequencing. Techniques like gel electrophoresis, spectroscopy and fluorometric analysis are used for assessment. Nanodrop and Qubit are two commercially available spectroscopy and fluoroscopy instruments available, respectively.
Nanodrop can reveal the quality and quantity of the DNA whereas Qubit can also reveal the quantity of DNA. As both factors are considered, Nanodrop light is the best choice for qualitative and quantitative evaluation.
Related article: Qubit vs Nanodrop Light.
DNA integrity is also taken into account as well. Isolate can be run on an electrophoresis gel to determine the quality, integrity and degradation of DNA if any. High molecular weight and intact DNA is sent to the next step. Capillary electrophoresis is also another alternative option for conventional gel electrophoresis.
DNA library preparation:
The whole genome can’t be sequenced in a single run. It’s huge and no machine can do that. So what we have to do is that we prepare the library of fragments for the sequencer to understand.
Library preparation has sub-steps like– fragmentation, end repair, adaptor ligation, amplification and purification. Better to understand every step separately.
1. DNA fragmentation:
It is a process to cut DNA into smaller, random and suitable-sized fragments. In fact, the read accuracy of sequencing highly depends on the fragment sizes. Restriction digestion and physical lysis are two common ways of DNA fragmentation.
It’s notable to know that the fragment size depends on the sequencing technology selected. For example, Illumina genomic sequencing platform ideally requires ~800 bp fragment libraries. The type of DNA sample and sequencing platform are two factors to choose the fragmentation technique.
2. End repair:
Now we have DNA fragments of similar sizes but will have uneven and damaged ends too. To ligate known adapter sequences to our DNA ends, we need to repair them and generate blunt ends. Adapters can only ligate if blunt end fragments are there.
T4 DNA polymerase and E Coli DNA polymerase Klenow fragment are two polymerase enzymes used for end repair. What any of these polymerases do is, perform exonuclease and polymerase activity, simultaneously.
3’ to 5’ exonuclease action removes 3’ overhangs while polymerase adds 5’ overhangs, both in combination generate sticky ends.
3. Adapter ligation:
Adapters are known and short DNA sequences, are allowed to bind with the fragments. Adapter sequences work as a unique barcode having known information. Each adaptor-specific complementary sequence is located in the sequencer flow cell.
It allows correct hybridization to occur and initiates the sequencing reaction. Adapters are commercially available and ligated using a ligase enzyme. The library preparation steps are complete here.
4. PCR amplification:
This step is known as library enrichment and is optional. When dealing with metagenomic DNA samples, having a low number of DNA copies for microbes, every fragment from the library is amplified.
An increment in the number of copies highly increases sequencing resolution, coverage and final read quality. However, it’s important to know that more copies may interfere with the results too. So library enrichment/amplification is performed if only required.
Library purification:
As aforesaid, Next-generation sequencing, like shotgun sequencing, is a highly sensitive technique, so prior to sequencing, the library must be purified, in order to remove unused adaptors, incomplete fragments, dNNTPs, and unamplified or partially amplified fragments.
Kits like a spin column or magnetic bead-based purification are recommended to achieve sequencing-level purified products. Each library is now quantified and applied for sequencing.
Our DNA sample is now ready for sequencing. In the last sequencing step, each nucleotide present in the sample is read by the machine and recorded. We have covered a comprehensive article on this topic, which you can read here: DNA sequencing: Definition, Steps, Process, Types, Applications and limitations.
Summary :
| What to do? | What not to do? |
| Check the quality, quantity and integrity of DNA. | Do not use unpurified DNA and libraries. |
| DNA samples for sequencing should have a purity of approx. ~1.80 (260/280 nm) and quantity >100 ng. | Avoid using colored mastermix. |
| Perform library preparation as advised by the manufacturer. | Avoid using chemical based kits for genomic DNA isolation. |
| Purify the DNA library prior to using and remove unused dNTPs, fragments, chemicals, etc. | |
| Check the integrity of the library using gel electrophoresis. | |
| Only use kit-based purification. | |
| Remote genomic DNA when dealing with plasmid DNA sequencing. |
Wrapping up:
In conclusion, preparing DNA for sequencing is a critical step in sequencing that requires careful attention to detail and utmost quality control. Accurate library preparation leads to better-quality sequencing reads and results.
Plenty of library preparation services are now available which saves time and cost for the experiments. Scientists can generate accurate and useful data only if DNA is prepared well.
I hope you like this article, Please share it and/or bookmark this page.