“A method to represent the evolutionary relationship between organisms, species or group of organisms is known as the phylogenetic tree.”
Simply put, it is the representation of the path between common ancestors to divergent descendants.
Every organism on earth is different at least genetically. Their DNA sequences vary. Also, the phenotype and visible characteristics of various organisms or species are dissimilar.
Based on the similarities and differences between the organisms and species the phylogenetic tree can be constructed.
Simply put, the phylogenetic tree, often known as a “tree of life” is constructed based on the evolutionary relationship between different organisms and/or species.
The DNA sequencing method nowadays is used so commonly to detect sequence variations in different species, it can detect even a single base change. And various species and organisms can be characterized effectively, though, visible characteristics are also taken into account whilst preparing the tree.
In the present article, we will understand what a phylogenetic tree is and how to construct it. Furthermore, we will also understand how to interpret different types of phylogenetic trees.
In the upcoming section of this article, we will try to explain to you some of the software used for phylogeny and a tree of life.
Quick recap:
A phylogenetic tree depicts the relatedness and differences between species. It can be constructed manually or using the software.
Rooted, unrooted, bifurcating and multifurcating are various types of trees popular among evolutionists.
Tip, branches and node are the common elements of a typical tree which is also denoted as taxons, taxa and common ancestor, respectively.
The entire group is included in a ‘clade’.
The phylogenetic tree is manufactured to measure the divergence between species, popularly, although the method is not quantitative.
The present method to separate species is not so accurate, on the downside.
Physical characteristics, DNA sequence data and protein sequence information are commonly used to make a tree.
All the species on earth are classified using the phylogeny known as the tree of life.
Key Topics:
What a phylogenetic tree is!
The phylogenetic tree shows the evolutionary relationship among organisms in terms of relatedness and differences. It is the best way to describe an organism and to know from where it was evolved, however, the technique is not 100% accurate to describe the alterations or divergences.
The term phylogenetics or related terminologies like phylogeny is derived from the Greek word ‘Phulon’ means ‘race’ or ‘lineage’ and ‘genesis’ means ‘origin’.
Though Charles Darvin had explained the concept of the tree of life, the first evidence of explaining relationships between different species was explained in the Elementary Geology book written by Edward Hitchcock.
Type of evolutionary tree:
Based on the various characteristics of the tree, various phylogenetic trees are classified into different groups such as a rooted tree, a non-rooted tree, a bifurcating tree, a multifurcating tree and an Enumerating tree.
Note that there is so much literature on these topics describing different types of trees but are most confusing, I am trying to explain it in simple language so that you can understand the concept behind it.
Rooted tree:
The rooted tree is described as a phylogenetic tree sharing the common ancestor on the node. Therefore the classification ends at one point usually on the node which is the common ancestor of all the branches of the tree.
unrooted tree:
Contrary to the rooted tree, the non-rooted tree doesn’t have a common ancestor. The unrooted phylogenetic tree is always prepared from the rooted tree by excluding the common ancestor or the node of the tree.
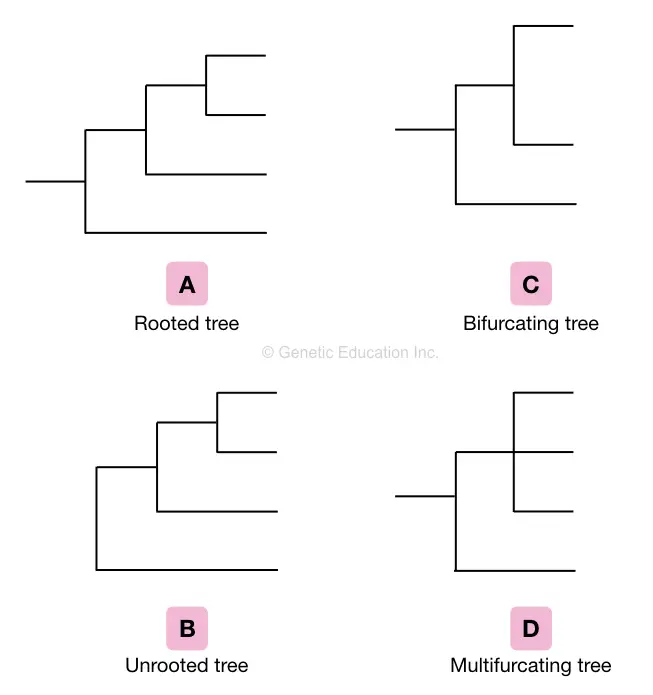
Bifurcating tree:
The phylogenetic tree only has two branches or we can say leaves are known as bifurcating trees. It is also classified in rooted bifurcation trees and unrooted bifurcating trees.
Multifurcating tree:
The multifurcating tree is described as having multiple branches on a single node. Again it is classified into a rooted multifurcating tree and an unrooted multifurcating tree.
It was believed that eukaryotic multicellular organisms evolved from primitive prokaryotes. There are several evolutionary forces that govern the process of evolution. Genetic factors are one of them.
Genetic polymorphism, gene flow and genotyping frequencies are some of them.
The phylogenetic tree is manufactured based on the phenotypic as well as genotypic variations and similarities between species.
This means when we will construct a phylogenetic tree, visible changes, as well as changes in the DNA sequences, are taken into account to achieve accuracy.
How to construct a phylogenetic tree?
Before, trying to construct a phylogenetic tree, first, let us understand several terminologies which are so useful in describing the tree. Here I have prepared a table for you. You can also correlate it with the explanation given below.
| Term | Explanation |
| Phylogeny | A method to construct a phylogenetic tree |
| Common ancestors | A group of organisms sharing the common feature with the decedent. |
| Taxon | An organism of the entire taxa |
| Taxa | A group of organisms from a species or from a different species. |
| Node | Nodes represent the common ancestors of Different taxons. |
| Sister group | Two or more taxon share the same node. |
| Outgroup | The taxon is outside the interest group or other than the common ancestor. |
| Clade | A clade is a group of all organisms from a common ancestor. |
First, the taxons are the organisms we are using to prepare a phylogenetic tree, taxons are denoted on the tip of the tree, shown in figure 1 below.
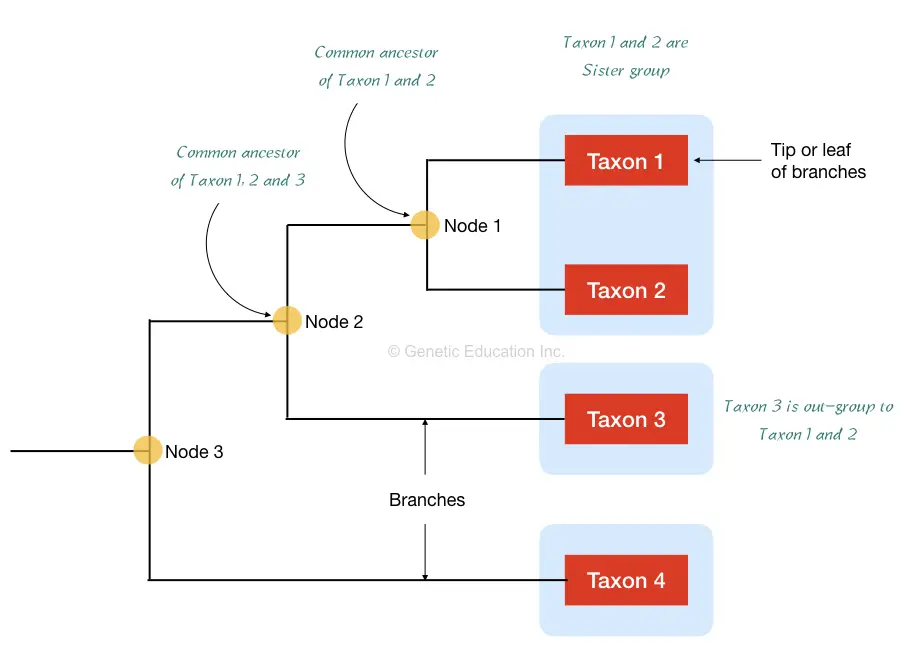
The Taxa is the group of all organisms who are sharing the common clade.
Here the taxon 1 and 2 are considered in the single taxa and sister group because of having a common ancestor.
Taxon 3 is considered as the outgroup to the entire taxa. Furthermore, node 1 is the common ancestor point for taxon 1 and 2 while node two is a common ancestor for node 1 and taxon 3.
From the above tree, we can say that the organism of taxon 1 and 2 are more similar in terms of phenotype and genetic sequences while the organism of taxon 3 is not so similar because it is not on the same lineage or node, though all three are sharing the common ancestor on node 2.
The node is also known as the branch point where different branches of organisms share a common ancestor. The 1, 2, 3 and 4 are different organisms or species of interest to study and located on branches.
The end of the phylogenetic tree is known as the “root” where the tree ended for the selected organisms. Notably, each node represents the most common recent ancestor of the group of species.
The phylogenetic tree can’t give us the quantitative data for the divergence of species. For instance, if we add another species to this tree as the Taxon 4, with a common clade 2, we can’t predict which of the two between the 1 & 2 and 3 & 4 are closely related.
Also, we can’t even predict how diverse the two sister groups are (sister group of 1 & 2 and sister groups 3 & 4).
Constructing a Phylogenetic tree by a software:
There are so many different software nowadays available to make a phylogenetic tree, some are complicated and some are simple to use. Here in the present section, I am explaining to you some of the simple methods to make a phylogenetic tree online.
You can start making a phylogenetic tree using the phylogeny tool available in EMBL-EBI.
Go google and open www.ebi.ac.uk.
Select the option services and choose the phylogeny option from the tools. Click on more tools or search it in the search box.
Select the simple phylogeny option.
You may see a three-step phylogeny process, step 1 enters your file of multiple sequence alignment or you can directly paste the data into the box given.
Now in the next step, select the options you want to study.
Tree formation: select any one of the clusters, distance matrix, or Nexus. I recommend making it default.
Distance: you can on or off the option to see the distance between the sequence or the species.
In the next option of “GAPS” select on or off as per your convenience whether or not to use the gap data.
In the cluster method section select UPGMA.
In step three select the ‘be notified’ option and submit the data.
Your tree will be constructed within a few minutes.
How to read a phylogenetic tree?
For a novice reading, a phylogenetic tree is yet another challenging task, it’s always a question from where to start!
The phylogenetic tree is unidirectional starting from a common ancestor to a divergence of species, though it is read and constructed from species to a common ancestor.
The species are located on the “tip” of the branches popularly known as the taxons or leaves of plants, whatever you want to call. If we read it, on a tree, every branch has different characteristics or DNA sequences.
Some of them end in the node where they are sharing the common ancestor. Commonly phylogenetic trees are rooted; end at the common node.
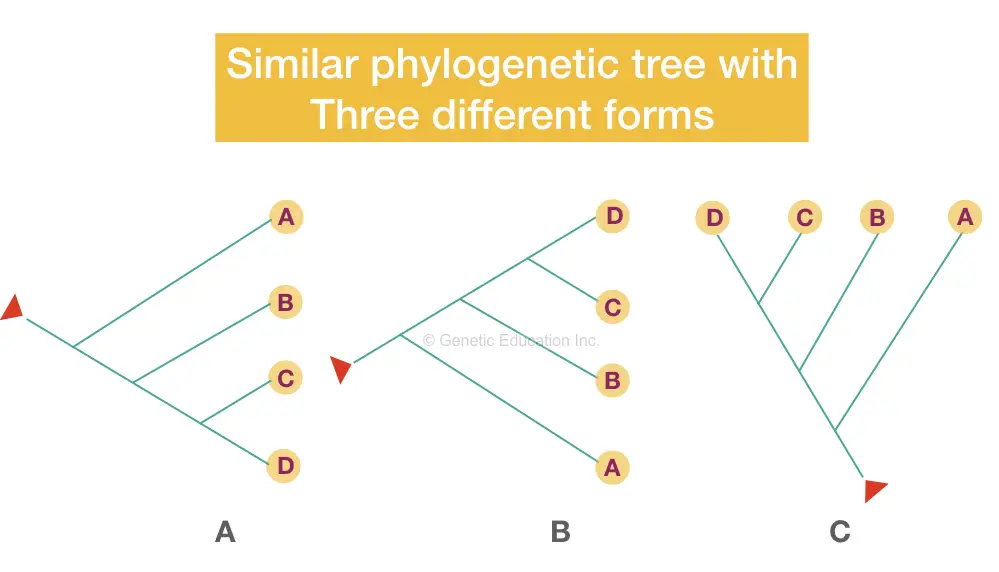
In phylogeny, the node is also known as a “clade” as well. Though there are so many different variations of the phylogenetic tree, every method of making a tree depicts the same type of information. Take a look at various trees shown in the figure below,
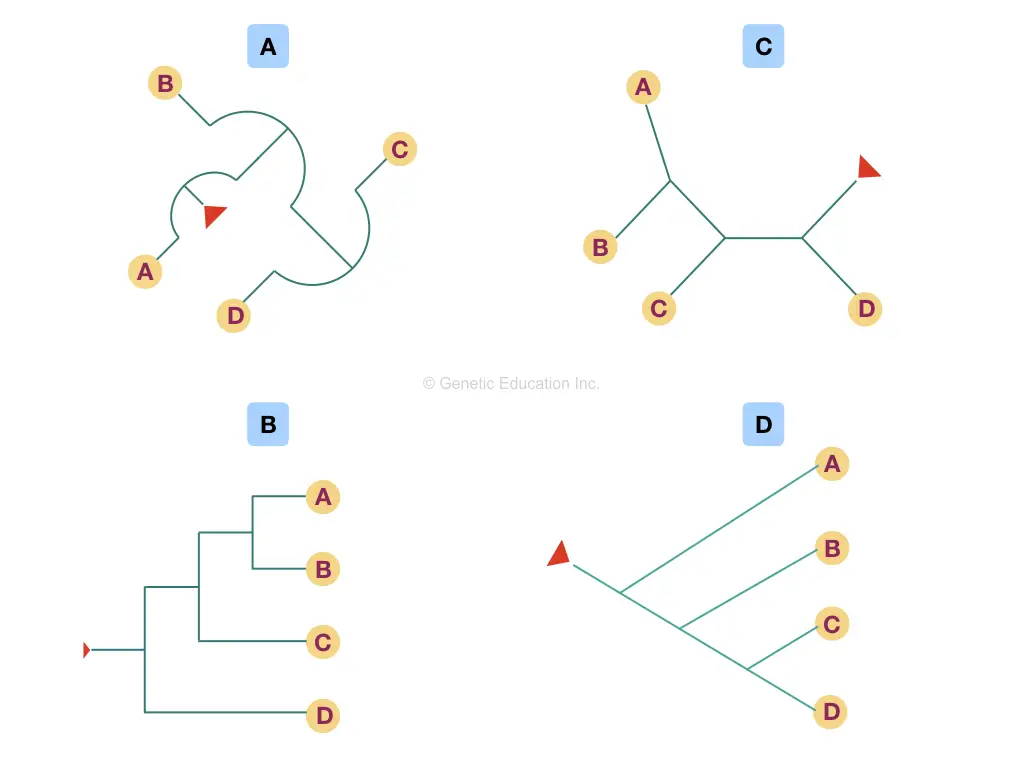
Keep in mind that whatever the shape, topology or structure of the tree is, it must have a common node if rooted and branched.
To read a tree, start with the tip of the branches and see where the branch ends (the node), based on that information you can depict or conclude which organism is nearer or closer and which are distantly related.
A phylogenetic tree of life:
The tree of life is the phylogeny of almost all organisms on the earth. The tree of life or the phylogenetic tree of life gives us all information on how different organisms are different from one another, the relative distance between two species and with whom they are similar.
Although for us it is very hard or near to impossible to make a tree of life covering all the organisms on the earth.
Here I am explaining a tree of life that I had found whilst doing research online.
Go to the online tool known as ITOL- interactive tree of life- www.itol.embl.de.
Select the options tree of life from the menu bar. You may see the image shown above.
Now what to do with the tree of life?
There are several options on the side of the window using which you can generate or obtain some information.
For example, you can first zoom in or zoom out the three because it is so big.
Using the ‘Basic’ option you can choose which type of tree you want to make!- circular, rooted or unrooted.
There are so many options there, try each one.
After that, you can denote or indicate or label a different group of species like you can choose the option Eukaryota, Bacteria or Archaea given below on the window. Even you can mark them in different colors.
Using the option ‘datasets’ you can give indications of genome size, publications and domains per genome data on the tree.
On the other side, you have the option to manually annotate, you can use it.
You can also share the tree of life on your social media platforms as well.
Applications of a phylogenetic tree:
- The phylogenetic tree is constructed to make an evolutionary link between various organisms. By doing so, we can get an idea about how and from whom different organisms are evolved.
- Also, it helps to classify organisms and species in different taxa and groups based on their DNA sequence and phenotypic similarities and differences.
- In addition to this, it is useful to study the force of evolution and characteristics of different organisms.
- It is applicable to study the events occurring during the course of evolution and to classify species based on the divergence of structure and function.
Limitations of a phylogenetic tree:
- Firstly, the manual process of constructing a phylogenetic tree is a tedious and time-consuming process. We need special software to create one!
- With this to construct a tree based on the DNA or protein sequence variations, a lot of lab work is needed prior to making it.
- For instance, if we want to construct a phylogenetic tree of 100 different organisms for a couple of genes. DNA extraction, amplification, and DNA sequencing are performed on all 100 samples prior to proceeding for phylogeny.
- The biggest limitation of the present technique is its accuracy. The Phylogeny methods to differentiate organisms or species are not so accurate.
- In addition to this, not all the characteristics can be taken into account for constructing a single tree, it will confuse things and will not give accurate divergence.
- Another limitation of the present technique is that we can’t quantify the divergence between two species. For instance, get back to example 1 at the starting of the article. Taxon 1 and 2 share common ancestor node 1 and taxon 3 and 4 share common ancestor node 2 but in which amount taxa 1 and 2 carry the diversity we don’t know.
- Furthermore, It is also difficult to explain which organism can be placed first in the sister group.
Conclusion:
The aim of constructing the tree of life or the phylogenetic tree is to relate or differentiate organisms or species based on their phenotypic and genotypic characteristics.
There are so many different paid and free online software that is now available to learn phylogeny. You can learn it there.
This article is just a comprehensive overview of phylogenetic trees. It gives information on the divergence of species and how it has occurred.
Sources:
Barry G. Hall, Building Phylogenetic Trees from Molecular Data with MEGA, Molecular Biology and Evolution, Volume 30, Issue 5, May 2013, Pages 1229–1235, https://doi.org/10.1093/molbev/mst012.