“Bisulfite sequencing is a technique for studying an epigenetic modification- DNA methylation. In this article, learn about the concept, technique, advantages and limitations of bisulfite sequencing.”
DNA Methylation is a prime and the most important epigenetic tag. When it occurs, it leads to gene silencing. It usually occurs at the cytosine base, thereby CpG-rich regions of the genome.
This epigenetic modification and many others, provides crucial insights into gene expression analysis and studies. Various PCR and DNA sequencing-based techniques are available to study epigenetic alterations.
However, it’s difficult to directly obtain sequence-level information regarding methylation. Meaning, methylation can not be directly studied, any sequencing technique can just tell us about the A, T, G and C but not about its elaborative structure.
So how is it possible to study DNA methylation using sequencing?
A technique known as bisulfite conversion is the gold standard method for methylation analysis. And when it is combined with sequencing or whole-genome sequencing, scientists can study sequence-level DNA methylation and collect related data.
In this article, I will particularly explain the process of bisulfite sequencing which helps students and researchers to understand the concept. And also demonstrates the complete process and pipelines for researchers and scientists.
Stay tuned.
Disclaimer: The content presented herein has been compiled from reputable, peer-reviewed sources and is presented in an easy-to-understand manner for better comprehension. A comprehensive list of sources is provided after the article for reference.
Key Topics:
What is bisulfite sequencing?
Bisulfite sequencing is a technique to evaluate the methylation pattern for a gene or whole genome using a sequencing approach. Methylation is usually reported in the gene promoter regions, particularly in CpG dinucleotides.
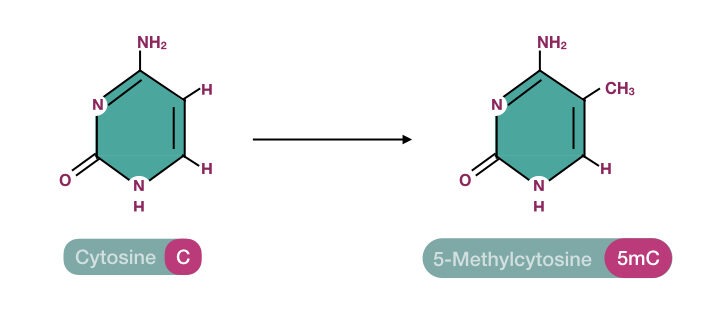
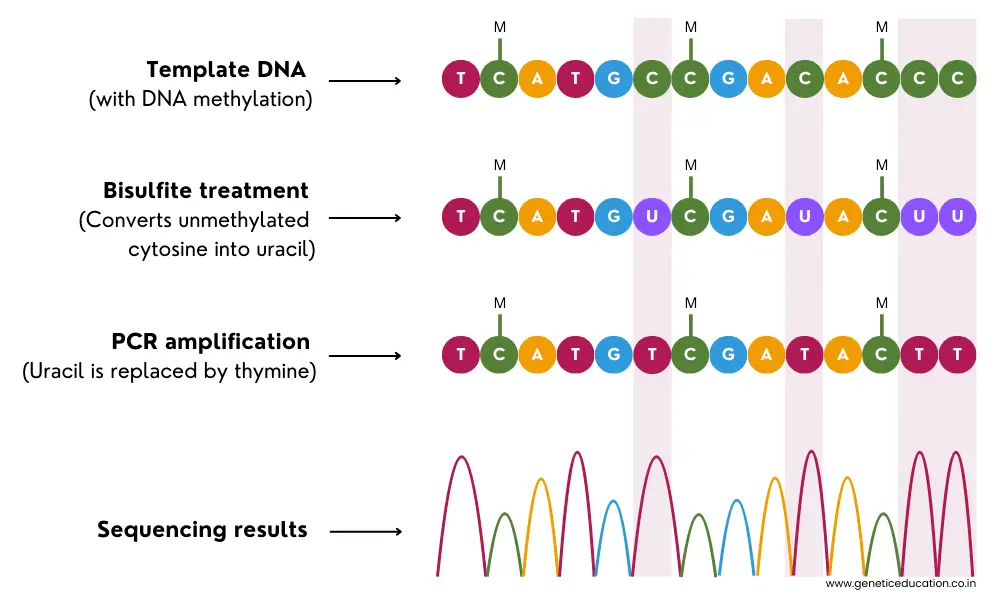
In this region, methylation occurs by the addition of a methyl group to the C5 carbon of the cytosine nucleotide. It converts the native cytosine base into 5-methylcytosine. Bisulfite conversion is the gold-standard technique to handle such operations.
After methylation, there are two types of cytosine bases present in a gene or genome, first the 5-methylcytosine (methylated) and native cytosine (unmethylated) bases.
Now, during the bisulfite treatment, sodium bisulfite converts the non-methylated (native) cytosine into uracil. Later on, PCR amplification converts the uracil into thymine which is detected during the sequencing read.
The graphical illustration is given in the image below.
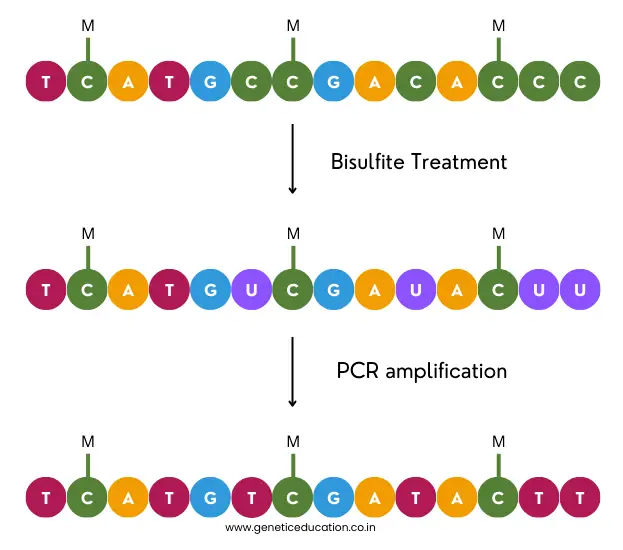
Upon completion of the sequencing run, the file generated by bisulfite treatment is compared with the reference or normal sequencing file to identify the ‘converted-thymines’ and to understand DNA methylation.
Let me tell you, all these analyses have been carried out by the software. The final output gives us quantitative methylation data.
How Is Bisulfite Sequencing Done?
Let’s dive deeper into the topic and learn the complete extensive process of how it’s done! Common steps in bisulfite sequencing are DNA isolation, bisulfite conversion, library preparation, PCR amplification, sequencing and analysis.
DNA isolation
Now, first! DNA is isolated from the sample. A ready-to-use DNA extraction kit or automated extraction is recommended to achieve excellent DNA purity and yield. Well-standardized manual protocols can also be used.
The extraction is followed by quality and quantity assessment. This gives us an idea about whether the extracted DNA can be used for sequencing or not. It’s important to note that during the bisulfite treatment, a huge portion of DNA can be lost.
Thus, a bit higher amount of DNA is required, particularly for whole genome bisulfite sequencing. Scientists suggest that 100nG to 2µG DNA can be used in WGBS. Notedly, too much DNA misleads the bisulfite treatment.
Ideally, follow the manufacturer’s instructions for genomic DNA isolation for bisulfite sequencing.
Bisulfite conversion
Conversion of cytosine to uracil (that is the bisulfite conversion) is a critical step in this entire process. We will explain the process elaboratively here, and cover all the information regarding bisulfite conversion in a separate article.
Ok so first, the denaturation process forms two single-stranded DNA from the double-stranded DNA. cytosines in the dsDNA prevent deamination and disallow conversion.
In the next step, the singular DNA is incubated with sodium bisulfite at adequate temperature. This converts all native and unmethylated cytosines into uracils.
Desalting and desulfonation have been performed in the last step to remove all the bisulfites and unconverted bases.
Note that rapid bisulfite conversion kits are now commercially available for whole-genome bisulfite conversion. These kits can yield a maximum amount of conversion with accuracy.
PCR amplification
In the next step, the converted DNA sample is amplified using a PCR protocol. Using the uracil-sensitive polymerase, the converted uracil bases are amplified into complementary thymine bases.
In the case of WGBS (Whole-Genome Bisulfite Sequencing), the genomic DNA is first fragmented, enriched and amplified using a bisulfite PCR kit. Now the sample is ready for library preparation.
Related article: Methylation Specific PCR- A Complete Technical Guide.
Library preparation
After PCR amplification, all the steps remain similar to normal sequencing. Fragment libraries are prepared. Important sequences such as sequence tags, indexes, sequencing primers, etc are added to fragments.
Before that, processes like end repairs and ligations are also performed. Library preparation may vary from platform to platform. Follow the manufacturer’s instructions.
Note that 200 to 500 bp long short fragments are prepared for the library. Meaning, that after this, the sample is processed using a short-read sequencing protocol.
Related article: Genomic DNA Library- Preparation and Applications.
Sequencing
The libraries are sequenced in a sequencer machine, and bases are read according to the chemistry of the machine. For instance, sequencing by synthesis reads bases using fluorescent synthesis chemistry whereas semiconductor chemistry reads bases using pH change, etc.
Sequencing run and accuracy depend on the sample type. A fragment or gene can be sequenced in a few hours while whole genome bisulfite sequencing can take a few hours to a day.
Analysis
Plenty of epigenetic and methylation sequencing analysis platforms and bioinformatics pipelines are now available. This makes the whole analysis part easy. Usually, each software can perform qualitative as well as quantitative methylation analysis.
Put simply, the process is like this.
Each reads are aligned to generate a read cluster and the complete sequence.
In the next step, the bisulfite-treated sequence is matched with the reference or untreated sequence.
This gives us information about the thymine bases that are added after the bisulfite conversion. Meaning, that the cytosine call that we have in the sequence is methylated cytosine because we already converted our unmethylated cytosine into uracil and thymine.
Check this image to understand the scenario.
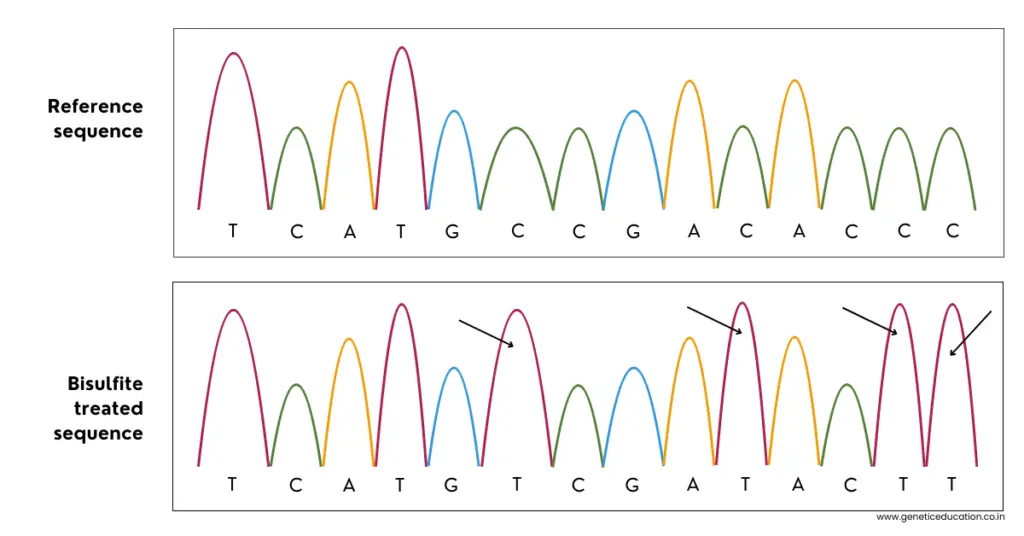
However, such manual analysis is difficult. The software performs all these analyses, counts methylation and gives us quantitative data. Now, let’s see some of the technical analysis that is usually needed.
Using alignment against the reference genome, as aforementioned, C-C matches and C-T mismatches can be studied. Noteworthy, only aligned reads are counted.
mC calling is another common analysis parameter used for methylation studies. Much like the variant calling in the case of normal sequencing; mC calling identifies the 5-methylcytosine regions from the genome.
mC calling is commonly employed for genome-wide methylation or methylome analysis. Read quality is the noteworthy factor for the present analysis. Meaning, that high-quality reads are considered for mC calling.
Genome-wide methylation analysis helps study the whole methylation pattern from the genome. Each methylated C is calculated using a computer algorithm and allows whole methylome preparation for the sample.
Lastly, some of the common whole-genome analyses such as sequencing depth and coverage are also considered for the preparation of the final report. Besides, differential methylation and methylation density analysis are also conducted.
Note that all these operations are based on computational and software analysis. Trained bioinformaticians are needed to conduct the analysis part.
Bisulfite Sequencing Techniques:
In this section of the article, I will explain various techniques that have been used to conduct bisulfite sequencing operations in various fields. Common techniques are Reduced Representation of Bisulfite Sequencing, Whole-Genome Bisulfite Sequencing, and Targeted Bisulfite Sequencing.
We’ll cover each technique individually.
Reduced Representation of Bisulfite Sequencing:
With a single nucleotide sequencing power, the present technique provides a reduced representation of whole genome bisulfite sequencing. Meaning, only highly methylated regions are sequenced, not the entire genome.
Although an additional restriction digestion step is involved in the present technique, RRBS has the power to sequence 80 to 85% of CpG islands. Meaning that it covers the majority of methylated promoter regions- approximately 85% of CpG dinucleotides.
So, only 1 to 3% of the genome can roughly be sequenced which saves time and cost.
Now, in the actual process, the Mspl restriction enzyme is used. Mspl, in particular, cleaves upstream to 5’CCGG3’ sequences and produces varied-sized fragments having the CCGG nucleotides.
Next, the RE-treated DNA is end-repaired, ligated with adaptors and used for library preparation. For library preparation, size selection is mandatory.
The sample is run on the agarose gel and 50 to 200 base pair long fragments are selected. The rest of the steps such as bisulfite conversion, PCR amplification, sequencing and analysis remain the same (as discussed in the above section).
| Advantages | Limitations |
| Cost-effective. | Can not study CpG lack and repetitive regions. |
| Selective sequencing of CGIs (CpG islands). | Can not cover the whole genome and thus can miss important regulatory elements. |
| High resolution and accurate analysis of 5mC. | Complex sample preparation. |
| Detection of quantitative differential methylation patterns of CGIs. | Labor intensive. |
| Scalable. | Require restriction digestion. |
The reason why RRBS is the best choice for methylome studies is because it greatly reduces the cost of the entire experiment and still provides the same results as the whole genome bisulfite sequencing provides.
Whole Genome Bisulfite Sequencing (WGBS)
Contrary to the RRBS, the WGBS sequencing covers the entire genome for sequencing. However, it doesn’t need additional restriction digestion steps. DNA extraction, bisulfite conversion, library preparation, sequencing and analysis are common steps in WGBS.
We have already discussed these steps in the above section, Please refer to it here: How does bisulfite sequencing work?
One of the reasons scientists perform WGBS is that it gives data other than the CpG regions of the genome. Moreover, methylation other than the promoter regions can also be studied using it.
| Advantages | Limitations |
| Covers entire genome-CpG, non-CpG, promoters, and other regulatory and gene regions. | Costly. |
| High-resolution single nucleotide methylation analysis. | Time-consuming and labor-intensive. |
| Quantitative differential methylation analysis. | Requires data storage, processing and analysis facilities. |
Read more: What is Genome Sequencing?- 3 Best Genome Sequencing Methods
Targeted Bisulfite Sequencing
TBS is a very feasible technique to study methylation. It focuses on a particular target which can be a sequence of interest, a gene, or any focused region of the genome. Unlike the RRBS, it doesn’t need any restriction digestion step.
The workflow is highly similar to the WGBS with an additional PCR amplification step. Common steps are DNA isolation, bisulfite conversion, PCR amplification, library preparation, sequencing and analysis.
In particular, PCR amplification is required during TBS to convert the uracil bases into thymine using the uracil-sensitive DNA polymerase.
| Advantages | Limitation |
| Focused and gene-specific methylation analysis. | Limited genome coverage. |
| Cost-effective. | Limited detection rate. |
| Reduced sequencing depth. | Low throughput. |
| Suitable for large-scale study. | |
| Efficient data analysis. | |
| Require less bioinformatics. |
Related article: What is Targeted Sequencing and How Does It Work?
Applications of Bisulfite Sequencing:
Now, coming to the important question of this article, how and where we can use methylation sequencing?
Gene expression studies:
One of the pivotal roles of the present assay is in gene expression and regulation studies. Scientists can investigate the role and expression of a single gene (targeted sequencing), many genes (whole-genome bisulfite sequencing) or various CpG-rich genomic regions.
Epigenetic investigations:
Bisulfite sequencing is thus crucial for epigenetic investigations. Such investigations include the complete genomic expression profile. By doing so, scientists can understand methylome dysregulation and its role in the development of various types of cancer.
Read more: Epigenetics 101: What is Epigenetics and How Does It Work?
DNA methylation studies:
To do so, scientists can conduct different analyses such as total methylation analysis, differential methylation analysis, whole methylome analysis or even help validate other methylation assays.
Scientists can compare methylation patterns between two biological samples, conditions or genomic regions which help understand the methylation’s role in various biological and physiological conditions.
Genomic imprinting studies:
Parent-of-origin-specific DNA methylation patterns for different imprinted genes can be studied with the present technique. Various diseases such as Prader-Willi and Angelman syndromes, different physiological conditions and behavior can be studied as well.
Related article: What is Genomic Imprinting? – Concept Explained.
Cancer genomic studies
Hyper and hypomethylation are both conditions that are linked with cancer. The tissue sample has been analyzed to study the methylation patterns and their link with the suspected cancer. Such investigations are routinely used for breast, oral, gastric and other types of cancer.
Aging studies:
Bisulfite sequencing is also employed for aging studies. Methylation alteration has been strongly linked with aging and premature aging, research suggests. Scientists utilize the present technique to investigate the methylation pattern to understand aging.
Besides, the present assay is also used in clinical diagnosis and research activities linked to various diseases and cellular activities.
Advantages of Bisulfite Sequencing:
Here are some of the advantages of the present technique.
- Sequence-specific methylation analysis.
- Single locus to genome-wide coverage.
- Qualitative and quantitative analysis of methylation.
- Detection of CpG as well as non-CpG methylation.
- High-resolution, accurate and sensitive epigenetic analysis.
- Gene-specific as well as differential methylation analysis.
Limitations of Bisulfite Sequencing:
Despite having immense use of the present assay in genetics and genomics, bisulfite sequencing has several limitations.
- It’s difficult to achieve complete genome-wide bisulfite treatment coverage. Several regions can not be treated accurately and are skipped from sequencing. Bisulfite conversion is a gold-standard technique but is insufficient for accurate analysis.
- Another limitation is also linked with the bisulfite treatment. It can degrade the DNA and create problems in sequencing. Meaning, bisulfite treatment can generate false-positive and false-negative results.
- It’s also noted that particularly for the targeted bisulfite sequencing in which we need PCR amplification, poor cytosine resolution by PCR also limits the present technique.
- The present assay needs extensive bioinformatics analysis pipelines, tools and expertise. This will increase analysis cost and time.
- Hence, bisulfite sequencing is a costly, labor-intensive, complex and time-consuming process.
Wrapping up:
DNA methylation is a common and important epigenetic tag for gene expression studies. Among many available methylation study assays, sequencing has been considered the most accurate one.
Bisulfite sequencing has a significant role in methylation study and is one of the important epigenetic sequencing methods. Various platforms offer bisulfite sequencing as per their chemistry. However, each platform needs bisulfite treatment to perform sequencing.
Still, data analysis is a challenging task for methylation studies. Besides, additional challenges are high cost, extensive bioinformatics and computational setups, etc.
We are planning a few videos and articles related to this topic. If this article is shared more and ranked decently on Google, we will add more content related to this topic.
I hope this article will help students, researchers and academicians in their learning.
Resources:
Li Y, Tollefsbol TO. DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol Biol. 2011;791:11-21. doi: 10.1007/978-1-61779-316-5_2.
Swarnaseetha Adusumalli, Mohd Feroz Mohd Omar, Richie Soong, Touati Benoukraf, Methodological aspects of whole-genome bisulfite sequencing analysis, Briefings in Bioinformatics, Volume 16, Issue 3, May 2015, Pages 369–379, https://doi.org/10.1093/bib/bbu016.
Darst RP, Pardo CE, Ai L, Brown KD, Kladde MP. Bisulfite sequencing of DNA. Curr Protoc Mol Biol. 2010 Jul;Chapter 7:Unit 7.9.1-17. doi: 10.1002/0471142727.mb0709s91.
Quentin Gouil, Andrew Keniry; Latest techniques to study DNA methylation. Essays Biochem 20 December 2019; 63 (6): 639–648. doi: https://doi.org/10.1042/EBC20190027.
Principle and workflow of whole-genome bisulfite sequencing by CD genomics.
Nakabayashi K, Yamamura M, Haseagawa K, Hata K. Reduced Representation Bisulfite Sequencing (RRBS). Methods Mol Biol. 2023;2577:39-51. doi: 10.1007/978-1-0716-2724-2_3.