“Huntington’s disease is caused by mutations in the HTT gene inherited in an autosomal dominant pattern.”
In autosomal dominant disease conditions, only a single allele of a gene is sufficient to cause disease.
Huntington’s disease is a progressive neurological-brain disorder that causes cognitive problems, loss of thinking, and uncontrolled movement. Further, emotional problems are one of the factors in it.
Huntington’s disease is caused by a special type of mutation called triplet repeat explanation disorder or trinucleotide expansion disorder in which the expansion of unusual triple repeat codon causes disease condition.
Especially, the CpG island of a gene is responsible for triplet repeat expansion disorder. In the case of Huntington’s disease, an abnormal expansion of the CAG triplet codon causes the neurodegenerative disorder.
The condition of the disease is based on the length of the CAG repeat in the HTT gene. The repeat length of more than ~40 causes the full penetrance of the disease in which the severity of it is very extreme.
The number of repeats is directly proportional to the severity of the disease. This phenomenon is known as anticipation. Interestingly, Huntington’s disease is previously known as “dancing disorder” or “epidemic dance” (because of the dancing-like movement and muscle activity of a patient).
The disease was first reported by George Huntington in 1872. The global prevalence of it is 1 in 10,000, however, it is most prevalent in the caucasian population.
The CAG triplet encodes for the amino acid glutamine thus the disease is often known as polyQ tract disorder or polyglutamine disorder. The number of the repeat and the severity of the disease is shown in the table below,
Table 1:
| Number of CAG repeats | Disease status |
| <27 | Normal |
| ~27 to 35 | Premutation (at risk of developing the disease) |
| 35- 39 | Premutation at high risk |
| >39 | Full mutation |
| 40- 50 | Adult-onset of the disease |
| >60 | Juveline form of the disease |
The HTT gene encodes for the huntingtin protein which supposes to function in neurons development in the brain. The gene is located on chromosome 4 at the short “p” arm at 16.3 (4p16.3).
Although the gene ‘huntignin’ is present in many tissues in our body, the highest activity of it is reported in the brain cells.
The structure of a gene is interesting and different from other genes as it contains the major portion of cytosine- adenine and guanine-rich region called trinucleotides.
The HTT gene also known as the IT15 gene is located on chromosome 4 while the CAG repeat is located on the 5’ untranslated region of it. Other triplet repeat expansion disorders are given in the table below,
Table 2:
| Disease | Gene | Location | Repeat | Normal range | Full mutation |
| Huntington Disease | IT15 | Exon 1 | CAG | 6-29 | 38-180 |
| DRPL | ATN1 | Exon 5 | CAG | 6-35 | 49-88 |
| SCA1 | ATXN1 | Exon 8 | CAG | 6-39 | 41-83 |
| SCA2 | ATXN2 | Exon 1 | CAG | <31 | 32-200 |
| SCA3 | ATXN3 | Exon 8 | CAG | 12-40 | 52-86 |
| SCA6 | CACNA1A | Exon 47 | CAG | <18 | 20-33 |
| SCA7 | ATXN7 | Exon 3 | CAG | 4-17 | 36- >460 |
| SCA17 | TBP | Exon 3 | CAG | 25-42 | 45-66 |
| SMBA | AR | Exon 3 | CAG | 13-31 | >40 |
| Synpolydactyly II | HOXD 13 | Exon 1 | GCG | 1-15 | >21 |
| Cleidocranial dysplasia | RUNX2 | Exon1 | GCG | 2-17 | >26 |
| HFGS | HOXA13 | Exon 1 | GCG | 12 | >17 |
| XLMR | ARX | Exon 2 | GCG | 16 | >17 |
| OPMD | PABPN1 | Exon 1 | GCG | 10 | >11 |
As we discussed above, the CAG repeats located one after another are responsible for Huntington’s disease.
Key Topics:
Symptoms of Huntington’s disease:
- Uncontrolled body movements
- Huntington’s chorea- involuntary jerking movements.
- Dystonia- rigid muscle
- In-coordinative movements
- Speech and swallowing problems
- Depression- anxiety- anger- apathy
- Uncontrolled emotions
- Learning and memory problem
- Slow or abnormal eye movements
- Frequent thoughts of suicide, death and dying.
- Difficulty on focusing on particular tasks, prioritizing and organizing.
- Lack of impulse control
- Insomnia
- Energy loss and fatigue
- Seizure
How is Huntington’s disease inherited?
Huntington’s disease is an autosomal dominant genetic disorder in which the mutation in a single dominant allele of the HTT gene is capable of causing the disease.
Related article: 100 Common Genetic Disorders.
Two alleles of the HTT gene are located on two different 4 numbers of chromosomes. If a single mutated allele is present on chromosome number 4, the individual suffers from the disease.
If one of the two parents is affected, the chance of the inheritance of the disease is 50% in offspring.
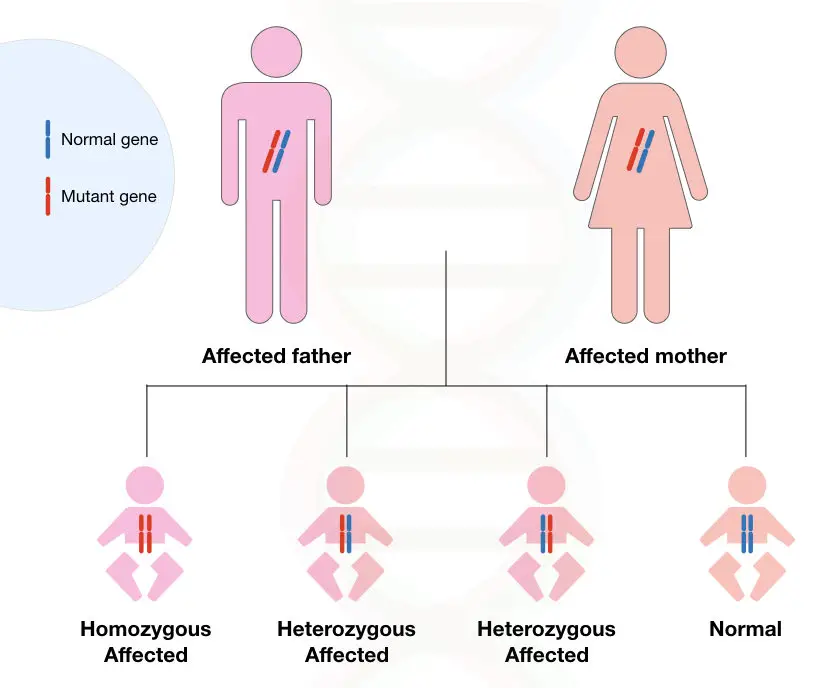
See image 1 below:

Two different conditions of Huntington’s disease are shown in the figures below, If both the parents carry a single dominant allele, the chance of occurrence of the disease is 75%.
In another condition, if single parents carry two dominant alleles, the occurrence of the disease in progenies is 100%.
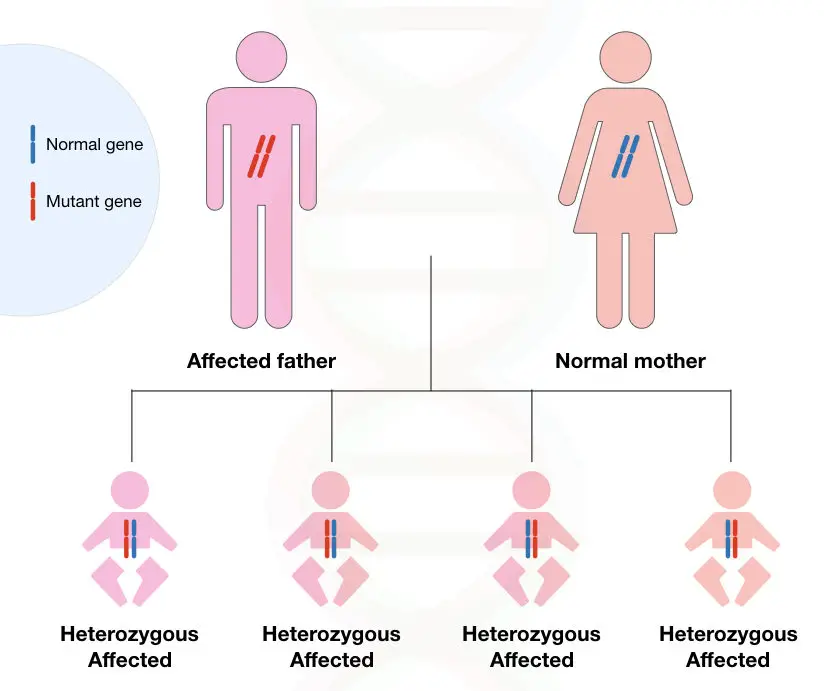
Although the disease is often characterized under non-mendelian inheritance (due to the role of triplet repeats in the disease), still the dominant allele is capable of causing full mutation.
Javelina Huntington’s disease occurs in childhood while adult-onset Huntington’s disease occurs in either thirties or forties of a person. However, the juvenile form of the disease is very rare and the number of triplet repeats in full mutation is more than 60.
Huntington’s disease facts:
Anticipation- the process of anticipation plays an important role in the development of the disease which is observed rarely in other disorders. the severity of the disease is proportional to the number of repeats present in a gene is called anticipation.
Dancing disease– the disease is also known as a dancing disease due to imbalanced and in-coordinative body movement.
Occurrence- the occurrence of the disease reported since the 16th century.
Mutation- the disease originated due to an unusual mutation of triplet repeat expansion in which a triplet CAG codon (which encodes glutamine protein) expanded abnormally.
Conclusion:
Genetic testing is one of the best methods for knowing “how is Huntington’s disease inherited.” The larger the amplicon band in a gel indicates a higher number of repeats in a sample. An autosomal dominant- single gene Huntington’s disease is one of the commonest motor and neurodegenerative disorders.
Nowadays, Huntington’s disease can be diagnosed prenatally by taking a DNA sample from amniotic fluid or chorionic villi.
Sources:
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5:40. Published 2010 Dec 20. doi:10.1186/1750-1172-5-40.
- Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci. 2016;18(1):91–98.
Quiz:
(A) The location of the IT15 gene
- chromosomes 1
- chromosome 4
- chromosome X
- chromosome 2
(B) The inheritance pattern of Huntington’s disease-
- Autosomal dominant
- Autosomal recessive
- X linked dominant
- X linked recessive
(C) Triple repeat in Huntington’s disease is
- GCG
- GAA
- CAG
- GAC
Answers:(A): 2- chromosome 4. (B): 1- Autosomal dominant. (C): 3- CAG.