PCR primers are short single-stranded, specific/ complementary DNA sequences, providing the free 3’ OH end for synthesis during the PCR reaction.
Polymerase Chain Reaction- PCR, is an in vitro DNA synthesis process much like replication. The technique relies on the use of different temperature gradients, unlike the replication which uses enzymes for performing various steps.
The PCR depends on the thermostable Taq DNA polymerase. It’s an enzyme that works even at a higher temperature which other enzymes can’t. Although the Taq needs a free 3’ OH end of DNA to polymerize.
In addition, it can’t add dNTPs directly to the denatured strand, it needs a short DNA strand using which it can start synthesis.
So for Taq DNA polymerase to work finely, it requires two things, a short, single DNA strand and a free 3’ OH group. Here the enzyme uses the single DNA strand to settle on while recognizing the OH end for adding nucleotides.
A primer is that thing! Put simply, it’s a single DNA strand, complementary to the target DNA and serves both the purposes that we have discussed so far. Notwithstanding, synthesizing artificial primers is indeed a tough job!
The present blog article contains information on primers, their definition and properties. Moreover, we will also discuss how to prepare it and different types of it. I also explain everything with an example.
So I hope this present article will add more value to your knowledge of PCR and DNA synthesis.
Stay tuned,
Key Topics:
What is a PCR primer?
Primer is a short DNA sequence used in the PCR.
PCR has true value in genetic science, used in many diverse fields like genetics, biotechnology, microbial genetics, environmental science and population genetics. It has a basic function to synthesize DNA, we know it as “DNA amplification.”
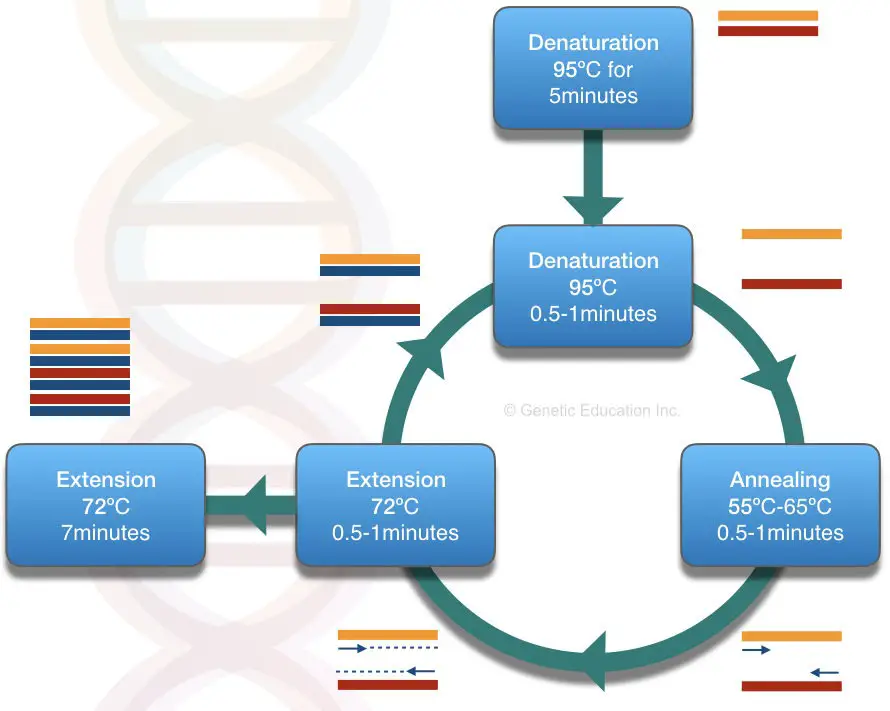
Put simply, the technique is completed in three steps; Denaturation, Annealing and Extension. Each serves, separating two single DNA strands, primer annealing to the target and elongation or synthesis, respectively.
It has basic ingredients like dNTPs, Taq DNA polymerase, reaction buffer, Primers, Template DNA and nuclease-free water. To learn more, read this complete article:
This present article basically focuses on the Primers only. The figure below shows the compelet process of PCR.
Definition:
A primer set consists of short, single-stranded, target-specific DNA sequences, provides the 3’ OH end and facilitates DNA synthesis during PCR.
Forward and Reverse Primers:
A typical single PCR reaction consisting of a pair of primers known as a forward primer and reverse primers, each synthesizes an individual target.
Forward and reverse primers ideally have the same length, however, aren’t complementary to each other. The forward primer binds to the extreme end of the one target at 5’ P while the reverse primer binds to another end of the 5’ P of another end.
Unlike the Replication process, the primer used in the PCR is DNA primers because of a couple of reasons,
- DNA primers are more temperature stable than RNA primers.
- As the polymerization process is unidirectional, RNA primers can’t be removed after the completion of the reaction.
- DNA primers are easy to synthesize and use in comparison to RNA primers.
- We are amplifying DNA not RNA so ideally it’s recommended to use DNA primers.
Note that the polymerase applied in the replication can have the power to remove the RNA primers after completion of the reaction.
Purpose of using primers:
The purpose of using primers in PCR is to facilitate DNA synthesis. It provides the free end and starting point for the polymerase.
Function of PCR primers:
The key function primer has is to help to copy the DNA. As we said, the single-strand provides the 3’ OH end and a catalytic site for Taq DNA polymerase to work.
Once the Taq finds the primer DNA function and recognizes the 3’ end, it starts adding nucleotides to the strand.
While we are diving in-depth into the topic, first we have to understand the temperature setup of the PCR. Among the three steps, primers get in action during the annealing step.
The annealing step has a temperature between 50 to 68ºC. A temperature at which the primer finds its complementary location and binds there is known as annealing temperature.
The annealing temperature is decided from the melting temperature of the template which is a temperature required to denature half of the template. To learn more, probably this article will help,
Incorrect annealing temperature results in non-specific binding and amplification, it varies among different primers.
Properties of PCR primers:
The ideal PCR primer should have several properties or we can say criteria in which it should fit. It relies on,
- The length of the primer
- The GC content
- Annealing temperature
- Complementation of each primer
- Start and end of primers
- Hairpin formation in primers.
Let us discuss one by one each property,
Length of primer:
The length of the primer is very crucial for PCR. Longer primers lead to non-specific amplification whereas shorter primers can’t do amplification. So it must be as per the requirement of the template.
The ideal length of the primer is between 18 to 25 (some say it is between 18 to 25) nucleotides. A ~20 nucleotides long primer gives the best results in PCR.
Primers <18 nucleotides have poor amplification affinity while primers >25 nucleotides compromise the reaction and give non-specific results.
In addition to this, each primer (forward and reverse) should have nearly the same size. Differences between a forward and reverse primer length alter the annealing temperature.
So both primers should have almost similar annealing temperatures.
GC content of primers:
Yet another crucial factor that has a direct impact on amplification is the GC content of primers. The GC content is the number of Guanine and Cytosine bases in a given DNA sequence.
G and C make stronger bonds as have three hydrogen bonds between them. And hence it needs more energy to break. Here the number of G and C nucleotides increases; it increases the required annealing temperature gradually.
The ideal primer should have GC content between 40% to 60%, although 45% is the best number for achieving excellent amplification. Meaning 8 to 10 GC nucleotides are tolerable in an ideal primer.
The higher GC in primer elevates chances of non-specific amplification as the G or C bases have a higher tendency to mismatch. Moreover, it spikes up the annealing temperature unnecessarily.
If summarizing things, to avoid complexity in the PCR experiment, choose the primer with 45 to 50% GC content.
Annealing temperature:
The primer anneals with the target location, on complementary sequence only, when it reaches its exact annealing temperature. Importantly, this parameter is as important as the primer length or GC content.
The annealing temperature should be 5ºC lower than the melting temperature. The melting temperature of the primer is calculated using the formula below,
Tm= 4 (G + C) + 2 (A + T)
An ideal annealing temperature of the primer is ranging between 50ºC to 62ºC. However, sometimes assay needs an annealing temperature >65ºC, for instance, more GC-rich templates, longer amplicons and high GC-rich primer.
Some say it should be between 55ºC to 65ºC. In that case, the assay requires additional optimization steps. I personally recommended the annealing temperature near ~57ºC (+/- 2).
Too high and too low annealing temperatures cause no amplification and non-specific amplification, respectively. If you wish to learn more on present topics, we have written an in-depth separate article. The link is given above.
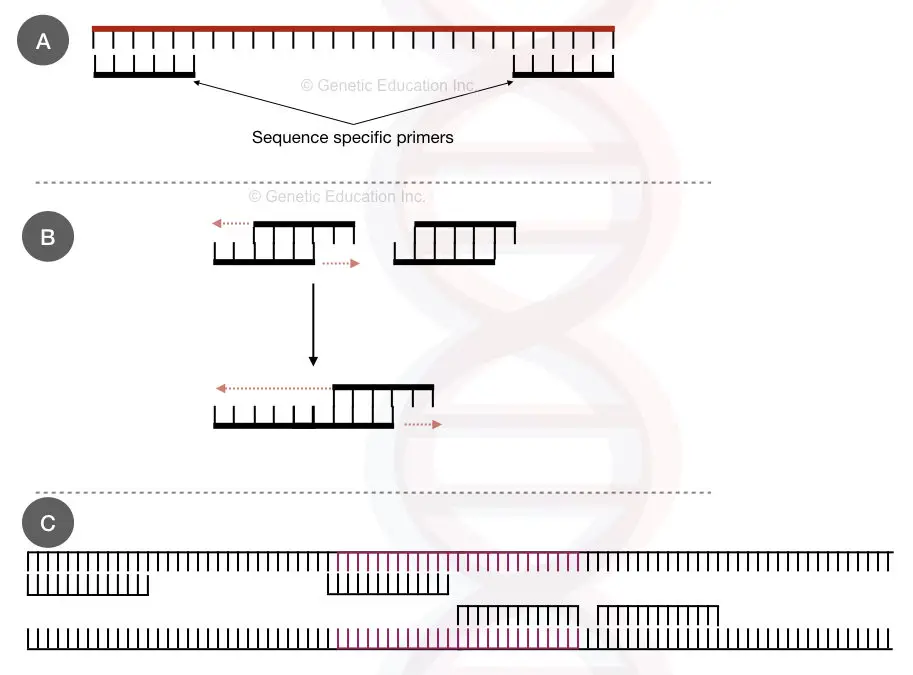
The complementary region between primers:
Every primer must have a unique sequence. For a single assay, or for any assay, the forward and reverse primer do not match with each other, that’s important.
If both have a few complementary bases, they annealing with each other and form primer-dimers instead of PCR products. When it comes to the “GC part” having a higher mismatch tendency only 5 to 6 complementary bases will amplify the primers. Meaning, we will get bands of primer-primer annealing, not our target amplicon.
Practically more than four complementary bases, lower annealing temperature and longer annealing step will produce more primer-dimers. The image below will make things clear.
See the figure below,
The melting temperature between each primer:
I think I have discussed something related to this topic somewhere in the article. Anyway what it tells us is that a primer pair should have a melting temperature within 5ºC of each other.
Understand that eventually, we are using a single annealing temperature for a pair of primers. So suppose if a forward primer and reverse primer have 50ºC and 65ºC annealing temperature, respectively, it makes things harder!
Meaning one premier’s activity will definitely compromise. So it is advisable that each primer should have a melting temperature difference within 5ºC.
Terminal nucleotides in the primers
When primers have repetitive bases or complementary bases within the sequence, it binds within and forms a hairpin-like structure. It makes the primer unavailable for amplification.
What this does do is that it makes the free 3’OH end unavailable for Taq DNA polymerase. So Ideally, a primer doesn’t have repetitive bases within. Also, note that, avoid the use of G/C bases at the 3’ OH end.
So in summary some of the important properties an ideal primer has are,
- Length between 18 to 25 nucleotides.
- Annealing temperature between 50ºC to 62ºC.
- GC content between 40 to 60%
- Melting temperature difference between forward and reverse primer between 5ºC.
- No repetitive and complementary bases.
- No complementary regions between forward and reverse primers.
If you are worried about how you can fulfill all these criteria in a single, let me tell you ‘yes’ it is hard doing manually. We do not have to worry about it, there are many primer design software that is now available as paid and free.
Now we will discuss how you can design a primer set using one of my favorite and popular primer design tools, the Primer3.
How to design PCR primers? A step-wise guide:
My PhD topics majorly cover many PCR experiments so I have designed many primers sets many times. I am sharing my process of how you can do it.
Firstly, Identify your template sequence. It is very important to know which gene or DNA fragment you want to amplify and obtain it from NCBI. It’s an easy task, if you don’t know I will make a separate blog post on it.
Primer synthesizing companies have their own primer design tools but I personally recommend using Primer 3, it’s free and open access, available to all. You can go to primer 3 from here: http://bioinfo.ut.ee/primer3-0.4.0/.
Actually, I think you should try it side by side in another tab. I will give you one sequence,
This is a beta-globin gene sequence (copy and paste it in a box of primer 3).
ACATTTGCTTCTGACACAACTGTGTTCACTAGCAACCTCAAACAGACACCATGGTGCATCTGACTCCTGAGGAGAAGTCTGCCGTTACTGCCCTGTGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGTTGGTATCAAGGTTACAAGACAGGTTTAAGGAGACCAATAGAAACTGGGCATGTGGAGACAGAGAAGACTCTTGGGTTTCTGATAGGCACTGACTCTCTCTGCCTATTGGTCTATTTTCCCACCCTTAGGCTGCTGGTGGTCTACCCTTGGACCCAGAGGTTCTTTGAGTCCTTTGGGGATCTGTCCACTCCTGATGCTGTTATGGGCAACCCTAAGGTGAAGGCTCATGGCAAGAAAGTGCTCGGTGCCTTTAGTGATGGCCTGGCTCACCTGGACAACCTCAAGGGCACCTTTGCCACACTGAGTGAGCTGCACTGTGACAAGCTGCACGTGGATCCTGAGAACTTCAGGGTGAGTCTATGGGACGCTTGATGTTTTCTTTCCCCTTCTTTTCTATGGTTAAGTTCATGTCATAGGAAGGGGATAAGTAACAGGGTACAGTTTAGAATGGGAAACAGACGAATGATTGCATCAGTGTGGAAGTCTCAGGATCGTTTTAGTTTCTTTTATTTGCTGTTCATAACAATTGTTTTCTTTTGTTTAATTCTTGCTTTCTTTTTTTTTCTTCTCCGCAATTTTTACTATTATACTTAATGCCTTAACATTGTGTATAACAAAAGGAAATATCTCTGAGATACATTAAGTAACTTAAAAAAAAACTTTACACAGTCTGCCTAGTACATTACTATTTGGAATATATGTGTGCTTATTTGCATATTCATAATCTCCCTACTTTATTTTCTTTTATTTTTAATTGATACATAATCATTATACATATTTATGGGTTAAAGTGTAATGTTTTAATATGTGTACACATATTGACCAAATCAGGGTAATTTTGCATTTGTAATTTTAAAAAATGCTTTCTTCTTTTAATATACTTTTTTGTTTATCTTATTTCTAATACTTTCCCTAATCTCTTTCTTTCAGGGCAATAATGATACAATGTATCATGCCTCTTTGCACCATTCTAAAGAATAACAGTGATAATTTCTGGGTTAAGGCAATAGCAATATCTCTGCATATAAATATTTCTGCATATAAATTGTAACTGATGTAAGAGGTTTCATATTGCTAATAGCAGCTACAATCCAGCTACCATTCTGCTTTTATTTTATGGTTGGGATAAGGCTGGATTATTCTGAGTCCAAGCTAGGCCCTTTTGCTAATCATGTTCATACCTCTTATCTTCCTCCCACAGCTCCTGGGCAACGTGCTGGTCTGTGTGCTGGCCCATCACTTTGGCAAAGAATTCACCCCACCAGTGCAGGCTGCCTATCAGAAAGTGGTGGCTGGTGTGGCTAATGCCCTGGCCCACAAGTATCACTAAGCTCGCTTTCTTGCTGTCCAATTTCTATTAAAGGTTCCTTTGTTCCCTAAGTCCAACTACTAAACTGGGGGATATTATGAAGGGCCTTGAGCATCTGGATTCTGCCTAATAAAAAACATTTATTTTCATTGC
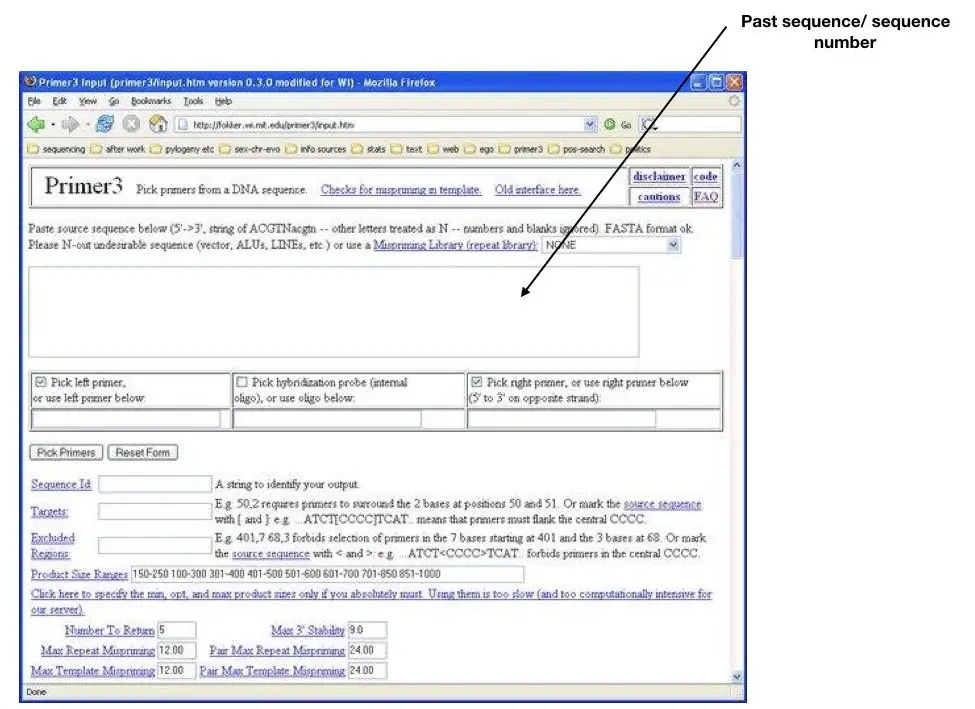
Now select the options for the forward primer and reverse primer shown as red arrows. Never select the option given in the middle (labeled as black) because we want to run the simple PCR hence we do not need a probe.
In the next step just for understanding read the information given on the primer-3 page, read the specification but do not click on any of the boxes because all the information is automatically set by default.
In the next step as shown in the figure, click on the “Pick primer” button and wait for the result.
The primer 3 output is shown in the figure (below). Analyze first the result window. You can see that the parameters like the length of the primer, GC content, annealing temperature and hairpin formation all are under the standard criteria.
Now take a look at the red line. The primer gives you 231bp fragment so when you run the PCR using these primers, your product should be 231 bp.
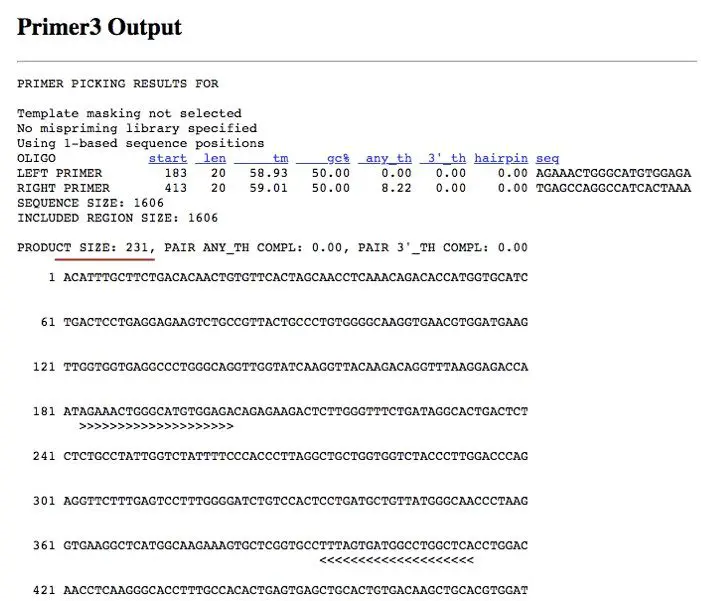
The arrows (>>>>>> and <<<<<<<) show the annealing site of the primer to your sequence. Additionally, the software gives you other pairs of possible primers at the bottom as shown in the figure (above).
Your primer is ready for the order. In the next step find out the company which gives service in your area. Send them the details or fill the online form of primer detail. While filling in details keep in mind to verify your sequence once.
If you made a mistake in a single base, you will not get the PCR result or false result.
Now when you receive your order, your primers will come in precipitate forms. Along with you will get a paper showing primer specifications from the company.
To let you understand, I am attaching my order here. Analyze it thoroughly.

The specification paper has all the information regarding the primer. It contains the yield at 260 nm OD, a sequence of primer, the yield of primer in microgram, the yield of primer in nano mol and other specifications as shown in the figure (above).
Our primer is in the form of a solid precipitate. We have to revive it for further use. Recall the PCR procedure, we need an approximately 10pmol primer for our PCR reaction.
The concentration of Primers in PCR
Let me explain how we can revive and prepare a working primer concentration. Suppose the given concentration of our primer is 29.1nM. When we add PCR-grade water of 291µl to the primer tube, the final concentration of our tube becomes 100pM/µl.
Do all the procedures in a sterile area now gently try to dissolve the primer in water. This concentration is our stock concentration of PCR primer.
To achieve a 10pmol final concentration for PCR reaction, take 1µl from the stock primer and add 9µl of water (again PCR grade) to it. Now our primer with 10 pM/µl concentration is read. We can use 1µl from this work.
In summary, the stepwise process of primer design is,
- Select a target sequence/gene or DNA.
- Obtain the sequence ID or sequence from the NCBI.
- Open the Primer 3 software.
- Past the sequence in the box.
- Click on Pick left and right primer.
- Click on the Pick primer button.
Optimizing PCR primer:
I love to write this section on every PCR-related article, because here I can share my personal experiences with you and how you can use them in your experiments. Some points might look so unimportant but trust me it makes a difference.
For example, add primers after, dNTPs and PCR reaction buffer but before the template. If you add a template first, primers start annealing even at non-complementary regions.
Use 10 pM, 1 µl each primer, but to avoid pipetting error take 1.2 µl in the reaction.
Make two or three aliquots of your stock primers.
Use a single press pipetting, do not double-press it.
Store primers at -20ºC.
Different types of PCR primers:
Conventional single-run PCR needs a single pair of primers. However, all reactions won’t be the same. We need optimization as per the assay requirement. In addition to this, the primer type may also vary depending upon the type of assay.
Here I have enlisted several types of Primers commonly used in the PCR.
Universal Primers:
Universal primers can be used in any type of reaction. For example, the RAPD primers are universal and can be used in phylogeny analysis of plant and plant DNA fingerprinting.
A single batch of 10 to 12 primer sets can amplify the DNA of any plant species and helps to establish a relation between them.
A novice can perform these types of experiments as all primers are generally processed at a single annealing temperature. In addition, it has great utility in cloning vector DNA amplification too.
Target specific primers:
A target-specific primer is employed to amplify a specific type of sequence, DNA or gene of interest. Meaning, it can’t amplify regions other than its complement, unlike the universal primers.
Mutagenesis and mutational studies rely on sequence-specific primers. Once can design these types of primers using the above-explained guide.
Degenerate primers:
Experts recommend using degenerate primers for microbial sample study and amplification. Technique: these types of primers have “kind of similar” sequences but not the exactly same.
It amplifies the same gene even in different organisms and hence can help in studying variations. Degenerative primers are less specific.
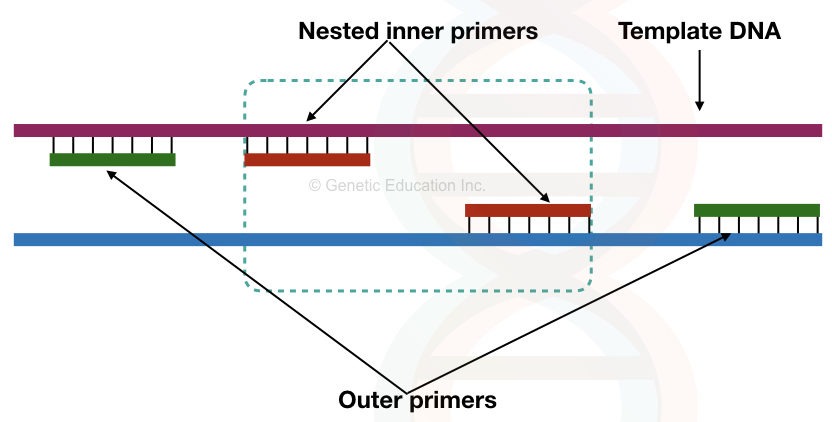
Nested primers:
Nested primers are a special type of primers used in the nested PCR reaction. Two sets of primers amplify a gene in which one set of primers is nested. This nested set of primer binds to the amplified product on the first set of primer.
The nested primer increases the chance of specific amplification by reducing the non-specific bindings.
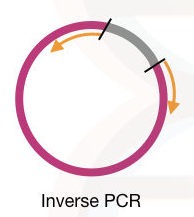
Inverse primers:
Inverse primers are the primer having the 3′ end outside of the template DNA, it amplifies the regions other than the target location. site-directed mutagenesis and in vitro mutagenesis studies rely on inverse primers.
It is also widely used in plasmid studies. See the figure below, how inverse primers amplify the DNA.
Conclusion:
Primers are the important ingredient of any PCR reaction, without it, we can’t predict the future of our amplicon. Although no special skills are needed to design a primer, high-end expertise is required to perform.
For long-term use of a primer, revive all the primer tubes in TE buffer and make different aliquots of the tubes. Store all tubes in -20°C. 10 pM/µl concentration is sufficient for the 25µl PCR reaction, excess concentration of primer results in dimers and non-specific binding.
I have covered all points on PCR primer design guidelines. You can comment below if any point is missing. Conclusively, using our PCR primer design guidelines, you can successfully obtain a result without any hindrance.

how are primers synthesized, please tell me full process of synthesis
how to get right amount of primers
loved the description, that helped me to better understand the concept. Thanks
I have a question please. The primers designed will not amplify the whole gene of interest but only a portion of it.
It depends whether you have designed primers covering the whole gene or some portion of a gene.
best article, i really liked. from theory to practical
everything is there.
Thank you
Thank you Naila.
loved the description
Thank you Dr saliha
it a good article for clearing the concepts
thank you haleema