“The site-directed mutagenesis is an artificial (in vitro) technique for introducing mutation or alteration into the target (known) DNA sequence.”
In simple words, we can say that
by using artificial techniques such as PCR a mutation or alteration can be inserted into a plasmid or target DNA.
A mutation is any sudden change in our DNA and is certainly harmful in most cases, that’s what we know about the mutation. But do you know we can induce artificial mutation? And use it for various purposes?
Those covers in artificial mutagenesis are often known as site-directed mutagenesis.
There are numerous applications of site-directed mutagenesis in gene therapy and gene-editing technology. It’s a kind of technique that tells you Yes or No about the assay. For instance, if a protein is correctly expressed or not; if a gene is working finely or not.
So basically scientists can do many things using the SDM- Site-Directed Mutagenesis. This article is more technical (not for a newbie) and comprises basic and advanced knowledge on site-directed mutagenesis, technique, applications and a lot more things!
Here in the present article, we are going to discuss different methods of introducing mutations or performing site-directed mutagenesis into the DNA. Why to do it and what is the purpose?
I hope this blog may increase your knowledge of practical genetics and help you in your future venture.
Stay tuned,
The content of the article is,
- Introduction to site-directed mutagenesis
- Importance of site-directed mutagenesis
- Different techniques for site-directed mutagenesis
- Direct/conventional PCR
- Inverse PCR
- Nested PCR
- Role of site-directed mutagenesis in CRISPR-CAS9
- Application of site-directed mutagenesis
- Conclusion
Key Topics:
What is site-directed mutagenesis?
Site-directed mutagenesis: Site: specific location into the genome, Directed– performed by artificial techniques, Mutagenesis– Introduction of mutation.
In the site-directed mutagenesis, at a specific location on the oligonucleotide sequence, the mutation is intentionally created and so named as Site-Directed Mutagenesis.
It’s often known as oligonucleotide-specific mutagenesis or site-specific mutagenesis. The purpose of introducing a mutation is to study the function of a gene. Usually carried out on plasmids.
The first site-directed mutagenesis (SDM) experiment was performed in the year 1974, in the laboratory of Charles Weissmann. The induced mutation was from GC to AT, however, the mutation was inserted randomly. Scientists from the Charles Weissmann university had incorporated a mutation and changed the GC nucleotide with AT.
Before the study of Charles Weissmann University, scientists were using artificial mutagens like chemical analogs and radiations to create a mutation in a sequence, those changes were random and non-specific.
In 1974, Michael Smith and Clyde Hutchison had performed site-directed mutagenesis using the primer extension method. They had achieved artificial mutagenesis at a specific location for the very first time in the history of genetics.
Types of mutagenesis:
Insertional and deletion mutagenesis are two common types, explained here,
Insertional mutagenesis:
Insertional mutagenesis is when a mutation is incorporated into the target gene. Here a few nucleotides are incorporated. Conventional PCR and nested PCR are two techniques that finely work for insertional mutagenesis.
Deletion mutagenesis
Deletion mutagenesis is when some bases are removed from the original/target DNA sequence. Inverse PCR is the best technique to produce deletion mutagenesis.
Principle of Site-directed mutagenesis
The basic principle of site-directed mutagenesis is simple: the primer possessing a specific mutation is artificially synthesized and used to amplify the gene of interest during Polymerase Chain reaction.
The DNA polymerase (high fidelity) extends the growing DNA strand bringing the new mutation. In the final step, DNA sequencing determines if the desired mutation is present in a gene of interest or not.
Transferring the GOI to a plasmid helps in downstream research and gene expression study. SNP (Single nucleotide polymorphism), deletion or insertion are common types of mutations employed to investigate SDM.
Related reads: Comparison Between DNA Primer And RNA Primer.
Techniques for site-directed mutagenesis:
The PCR has applications in detecting & studying mutations and introducing mutations at a specific location. The process used to screen a mutation is also applied to insert a mutation.
Several common and popular techniques used to do so are listed and explained here,
- Conventional PCR
- Nested PCR or primer extension
- Inverse PCR
Conventional PCR:
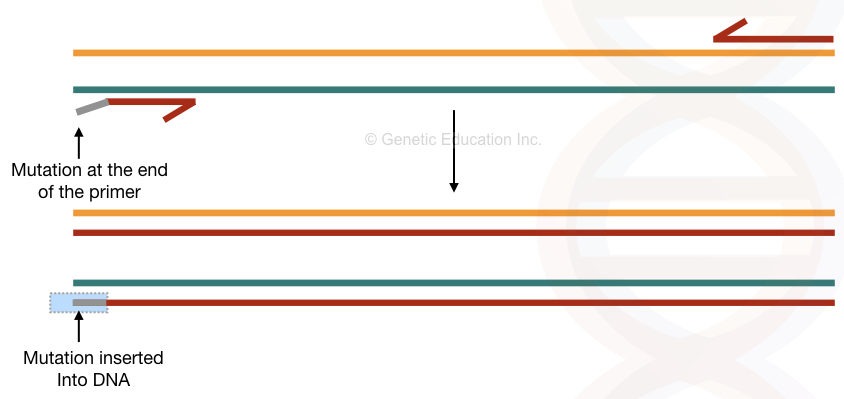
The conventional PCR method is one of the most popular and easy methods for site-directed mutagenesis. The setup is simple; a short, single-stranded oligonucleotide primer is designed in such a way that it possesses the mutation we wish to study.
Using the Taq DNA polymerase that lacks the exonuclease activity allows us to incorporate the mutation. As it can’t identify the mismatch during amplification it can’t remove it. If the mutation is minimal enough to incorporate, the primer binds to the DNA and amplifies.
Over a period of some amplification cycle, the DNA sequence having a mutation- the new one we incorporated replaces the original DNA sequence. The major recommendation for the conventional PCR-based mutagenesis is to insert mutant bases up to several limits at the 5′ end of the primer or in the middle of the primer.
From the 3′ end, the Taq polymerase expands the DNA. So the chance of reaction failure is higher there. Another limitation of the present method is that it carries mutant as well as normal/target DNA (mixed types of DNA) due to the presence of template DNA.
Hence the yield of the conventional PCR-based site-directed mutagenesis is lower. For more detail on conventional PCR, read the present article: A Complete Guide of the Polymerase Chain Reaction.
Noteworthy, the high fidelity DNA polymerase can’t work here, however, other assays need it. The Taq of the conventional PCR approach can alter 2 to 3 nucleotides per template.
The overview of the present method is shown in the figure below,
Primer extension or nested PCR:
In the primer extension, 2 sets of primers are used in which a single primer set is nested. Here the mechanism of carrying the mutation is the same, the mutation is introduced into the primer at one of the ends.
Usually at the 5’ end or in the middle.
Now let’s name the primers for convenience,
Let say the outer forward primer is 1 the outer reverse primer is 4
The inner forward primer is 3 and the inner reverse primer is 2.
The location of all four primers are shown in the figure below,
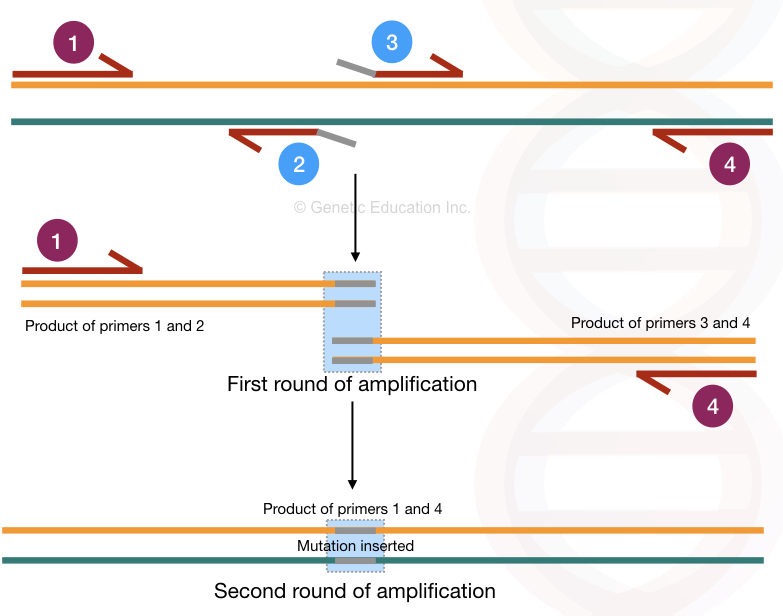
Here, the inner primers 2 and 3 contain the mutation/ mismatched bases at the end. The entire amplification process occurs in two rounds and so it generates two different amplification products.
In the first round of amplification, primer 1-2 and primer 3-4 create two different PCR products.
Primers 2 and 3 are mutant primer sequences therefore in two different PCR products, two different mutations are incorporated. In the second round of amplification, the primer 1 and 4 amplifies the entire DNA sequence having the desired mutation.
- Read more on nested PCR: What is nested PCR?
Inverse PCR:
In the inverse PCR, the primers amplify the fragment other than the target sequence. This means it amplifies in the reverse orientation.
The method is used for deletion mutation into the plasmid having a gene of our interest. The high fidelity DNA polymerase is used to do the amplification as well as to linearise the circular plasmid DNA.
After inserting mutation (meaning the “deletion”, we have introduced), 5’ phosphorylated end ligates linear DNA by ligase enzyme. And make the plasmid circular by intermolecular ligation.
The present method relies on the flanking region amplification thus it is used so often in deleting sequences from a plasmid. See the figure below,
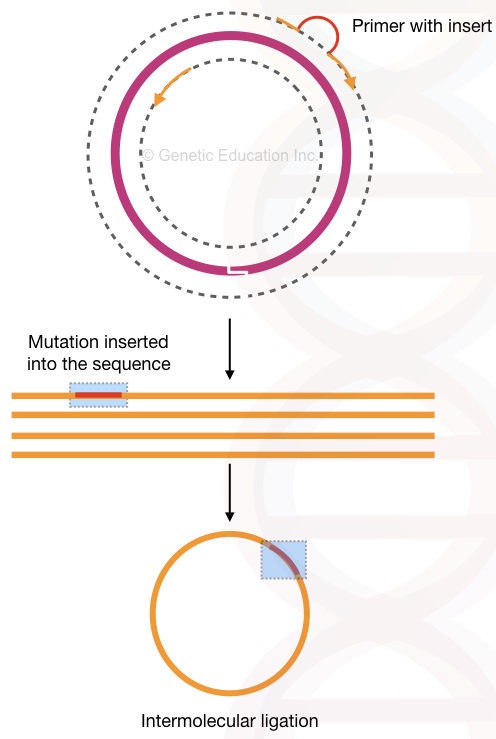
So technically, the inverse PCR technique can delete sequences from the gene of interest. We can delete many nucleotides through it. I have explained the process in the figure below. Although it can also do insertional mutagenesis by using a different assay.
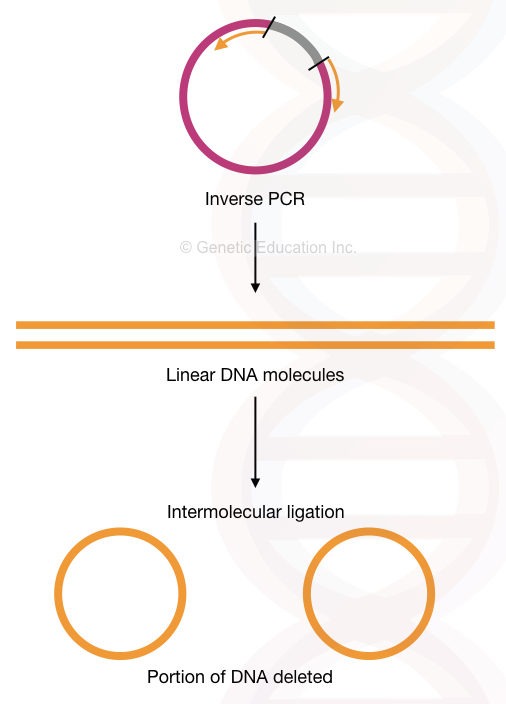
Read more about our inverse PCR: Inverse PCR: Principle, Procedure, Protocol and Applications
These three methods are most popular for site-directed mutagenesis. Now take a look at some of the components used in the PCR reaction, especially for the site-directed mutagenesis.
Template DNA:
Here, our template is always a circular plasmid DNA because we want to study the gene of our interest, present in a plasmid. Read more on plasmid: Plasmid DNA- Structure, Function, Isolation and Applications.
Notedly it uses other types of DNA when used for gene editing, gene manipulation and gene therapy experiments.
The concentration of the plasmid DNA must be between 0.1-1.0 ng/μl and it must be highly purified. Use a plasmid purification kit to purify the plasmid DNA. If the GC content of the plasmid is higher, use DMSO in the PCR reaction.
DNA polymerase:
The assay needs a different type of DNA polymerase having 5’ to 3’ polymerase activity for amplification and 3’ to 5’ exonuclease activity to increase the rate of fidelity and polymerization. Note that it’s essential that the polymerase lacks 5′ to 3′ exonuclease activity
The high fidelity DNA polymerase with 5′ to 3′ exonuclease activity removes the mismatched nucleotides while the 3′ to 5′ exonuclease activity increases the amplification specificity.
Technically, during the 3’ to 5’ exonuclease activity, the polymerase can’t go back and remove the target mutant (mismatched) nucleotides from the primer. And that’s to avoid 5′ to 3′ exonuclease activity here.
DNA polymerases such as Pfu, Vent, and Phusion facilitate a higher amplification rate in site-directed mutagenesis. The Taq DNA polymerase is only used in the conventional PCR-based method for introducing mutation.
Primers:
The technique needs primers that are totally different from conventional PCR primers. Here the mismatch or mutation is incorporated on the 5’ end. When the mutation is inserted in the middle, make sure to add 11 nucleotides complementary sequence on both sides.
Also, primers used in the site-directed mutagenesis do not have repetitive sequences and secondary structures.
3 base mismatch is tolerable in the ideal 22 to 25 nucleotide long primer. When the primer length increases, the specificity of the reaction decreases.
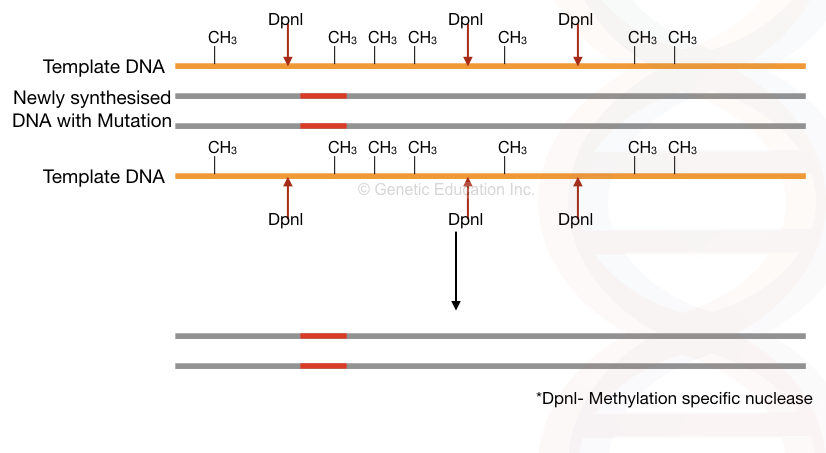
Nuclease:
Now, this is something very important.
We want to remove the template, unmutated DNA sequence from the assay right. The template DNA is natural DNA, it must contain a methyl group, which means, is methylated. While the newly formed DNA is unmethylated.
The methylation-specific endonuclease- a type of nuclease used here which cleaves only the methylated DNA. Consequently, it cleaves and removes methylated template DNA from the reaction. Only the mutant allele remains in the reaction. See the image,
Steps and procedure:
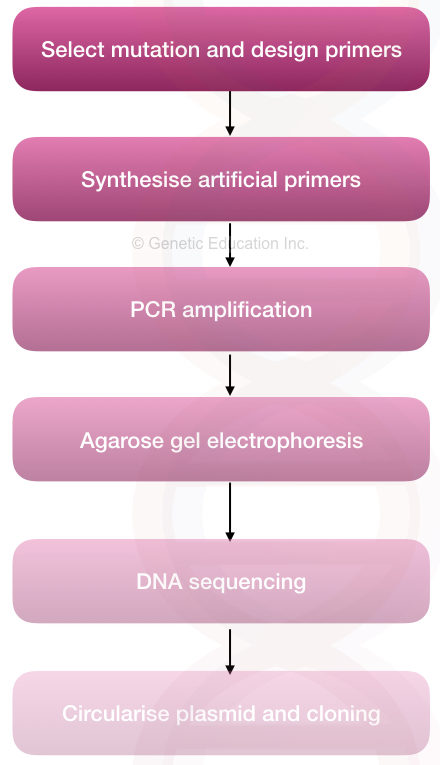
The whole process can be divided into a few steps,
Steps:
- Selecting a target and mutation to study
- Selecting a technique for SDM
- Designing, preparing and synthesizing primers
- Designing and performing PCR
- Validating results
- Validating alteration by DNA sequencing
- Downstream processing
Selecting a target and mutation to study:
The very first step in the mutagenesis process is to select the template DNA and mutation we wish to introduce. Several parameters prior to the experiment are evaluated such as template length, GC content, annealing temperature, template type, etc.
Besides, the mutation which will be introduced is examined carefully or evaluated for its possible results by computational analysis.
Selecting a technique for SDM:
In the next step, depending upon the type of template and the mutation, the technique is selected. A few techniques I have enlisted in the above segment. For example, the assay introduces a deletion in the target using the inverse PCR technique.
Designing, preparing and synthesizing primers:
Afterward, design the primers or set of primers depending upon the characteristics of the template DNA and the technique chosen. I have written an entire article on how to design primers, click the link and read it.
Send the primer sequence for synthesis and prepare the working stock solution of primers before PCR.
Designing and performing PCR
Next, design the PCR; what and in which amount you need ingredients. Estimate the number of PCR cycles required, the type of DNA polymerase and run time. Run the PCR accordingly.
Validating results:
Results of the experiment can be validated on the agarose gel. If dealing with the restriction digestion, use 3% gel otherwise use 1.8% gel. After validating the results, the samples are sent for DNA sequencing.
Note that care must be taken while preparing and running the agarose gel as it involves some harmful chemicals.
Validating alteration by DNA sequencing:
DNA sequencing allows one to investigate if the sequence carries the desired mutation or not. Sequencing examines the entire sequence of the altered and the native DNA sequence and relates both for comparative analysis.
DNA sequencing also validates the specificity of mutagenesis, meaning if mutagenesis occurred at a specific or random location. This step is very important in order to get success in downstream processing.
Downstream processing:
Once the mutagenesis is confirmed, the sample is processed for downstream assays like gene cloning, restriction digestion, gene editing, gene manipulation and gene expression study.
Protocol for site-directed mutagenesis:
The experimental setup is explained here:
- Design the primers to insert specific mutations (deletion, duplication, or SNP). Check all the primer parameters.
- Synthesis oligonucleotide primers artificially
- Isolating the plasmid DNA carrying a gene of interest for the study. Purify the template DNA if needed.
- Select one among the available techniques as per the requirement of the assay.
- Prepare a PCR reaction and perform PCR as per standard protocol.
- Validate the amplicons using the agarose gel electrophoresis.
- Purify the amplicon using a ready-to-use DNA purification kit.
- perform DNA sequencing to establish the mutagenesis results.
- Send the sample for cloning.
Key to success: Three important key hacks to success in mutagenesis study are the use of endonuclease, selecting and designing primers.
Applications:
The site-directed mutagenesis helps to improve the quality of the protein by removing harmful elements from it. These genetically modified proteins have high commercial values.
Let us justify this point. Suppose we don’t need some of the domains from a specific protein, for example, let say it is harmful. Using the inverse PCR technique, we can remove that particular domain-specific gene sequence from a target.
The tool has the capacity to study gene characteristics, gene properties, proteins encoded by a gene and post-transcriptional modification.
It also has applications in gene synthesis and gene-editing technology.
It can be utilized in gene cloning too.
By mutating the promoter or the regulatory regions of a gene, one can construct the map of the regulatory elements of a gene.
It is also useful in the screening of SNPs as well. In addition, the effect of the SNP can also be ruled out.
The best application of the site-directed mutagenesis is in screening restriction digestion, whether occurred or not. Two ways for doing it are; by inserting the restriction site or by removing the restriction site from the plasmid.
Removing or inserting the restriction site depends on the experiment requirement and both can be used for validating the target.
For example, when a restriction site is incorporated in a plasmid, near the gene or interest, it will show different bands when digested with a specific restriction enzyme following agarose gel electrophoresis.
Absence of restriction digestion bands indicating a failed reaction.
When we remove the restriction site from the plasmid, bands of digested products aren’t seen in a gel. In both cases, mutagenesis occurred successfully. Presence of restriction digestion bands indicating failed mutagenesis reaction. The image below says a thousand words!
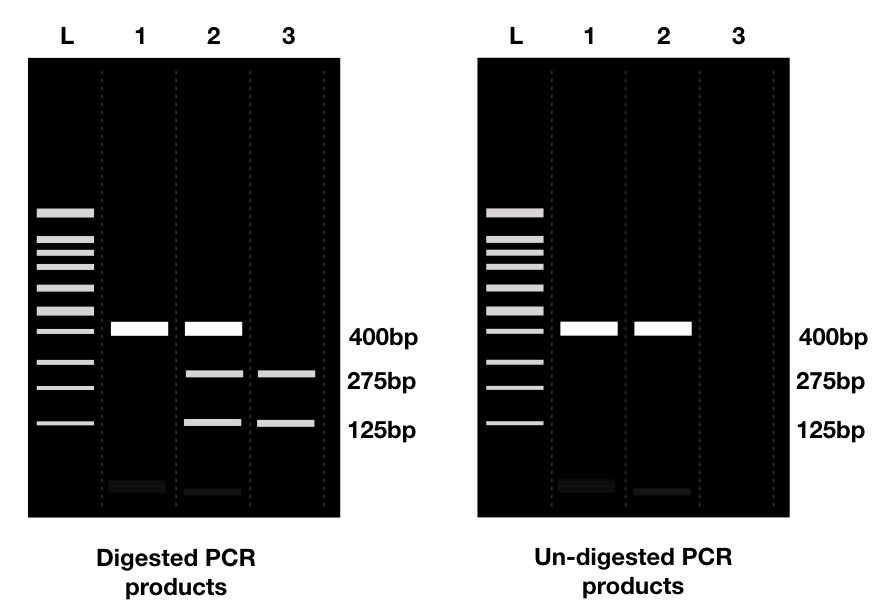
Removing a restriction site helps to differentiate a GOI from other sequences from the plasmid. Repeating digestion again after the SDM helps to know if it occurred correctly or not.
The technique helps to investigate the structure and function of a protein encoded by a specific gene.
When we introduce a mutation in a gene of interest, we suppress its activity. Phenotypic assay helps to distinguish the protein-related gene from the non-protein-related genes. Meaning, Genes’ phenotypic activity can be investigated.
The same technique of PCR-based mutagenesis has the power to investigate the structure and function of proteins related to specific genes.
For example, suppose a gene encodes one wild-type protein and it is required in the metabolic pathway of one organism. By mutating this protein-coding gene, the effect of the absence of that protein can be encountered.
Also, it has great utility in the gene-editing technique and gene manipulation. Through gene manipulation, the activity of a particular protein can be determined as well.
Besides these important applications, the method is often practiced to study protein-protein interaction, enzyme alteration, and enzyme silencing.
Role of site-directed mutagenesis in the CRISPR-CAS9:
Many ready-to-use mutagenesis kits exist in modern times for various purposes. One of the most important merits of site-directed mutagenesis is in gene editing, and CRISPR-CAS9.
One can introduce a point mutation in a model organism using both site-directed mutagenesis and CRISPR-CAS9.
Here, in the CRISPR-CAS9, the CAS9 is the nuclease, cleaves the DNA. Once it induces a double-stranded break, the mutation is inserted through the homologous-direct repair.
Ligase seals the nick. The newly altered gene is incorporated in a plasmid, incorporated in host cells, cultured and transformed. Read more on the homologous-direct repair (HDR): What is gene editing and CRISPR-CAS9?
Read related article: CRISPR-CAS9: step-by-step.
Advantages of SDM:
- The assay doesn’t need fancy methods of fluorescence chemistry. Simple PCR modifications like nested PCR, inverse PCR or conventional PCR can do the job effectively.
- The technique is simple, rapid and accurate.
- It enables us to study a gene or protein structure and function.
- It is cost-effective too.
Disadvantages:
- Several polymerases are more error-prone and thus can incorporate an undesirable mutation at the non-specific location.
- The technique is a bit complex when it comes to nested PCR or inverse PCR.
Conclusion:
Genetic experiments are lengthy, indeed but are worthwhile and that’s why researchers are constantly experimenting, finding new techniques and establishing new methods. Techniques of genetic sciences are certain sensitive too.
Huge experience and accurate working capabilities are required to perform. The SDM is one of those experiments.
Site-directed mutagenesis has its own importance in the field of gene editing and gene manipulation. It facilitates improvement in the wild-type genotype to produce a commercially important phenotype.
Notably, the use of nuclease is crucial to get success. site-directed mutagenesis is used in the knockout mice construction and gene knockout studies too.