“Metagenomics analysis is a technique to isolate, observe and study microbial DNA present in any environmental or biological sample and assess microbial diversity and functional variability within the existing microbial community.”
Download this article as an ebook: Metagenomics eBook.
Microorganisms have a significant impact on every living organism on Earth, directly or indirectly. Some microbes are useful, some are not, and a vast portion of them is still unknown. Microbes are present everywhere on Earth, ranging from the atmosphere to the deep sea, and they knowingly or unknowingly impact our environment and life.
Metagenomics is an interdisciplinary field of genetics. It includes cutting-edge, futuristic, state-of-art and high-throughput sequencing analysis for microbial studies. Such technologies are needed because of the flaws and limitations of conventional microbiology techniques. Here are some reasons.
Microbial culture, cultivation and microscopy are conventional methods to investigate microbes that are time-consuming, inefficient, and less accurate and unconventional. A Plethora of microbes is present in any sample that these techniques can’t study.
Conventional microbiology methods are prone to contamination, give false results and also, need human efforts to elucidate results.
Hence, traditional cultivation and culture-based microbiology techniques are inefficient for exploring the uncovered and unknown portion of the present plethora, which makes it practically impossible to identify every microbe present in any system.
Another problem we face with new-age 16S rRNA gene sequencing which only can determine taxonomic information and for high abundance microbes only. Yet again, a huge concentration of the microbial community and less abundant sequences remained un-studied.
Metagenomics solves both critical problems and allows us to investigate microbiomes effectively, with speed, accuracy, and precision. It provides an opportunity to study the vast portion of the microbial community that has remained unexplored and unknown.
With metagenomics, we can explore the microbes present in our gut, intestine or soil. For instance, a single gram of soil consists of 4000 to 5000 different species of microbes, while our intestines consist of 500 different types of bacteria. Metagenomics enables us to understand the diversity, abundance, and interaction of microbes in any system, making it a powerful tool in microbiology.
Let’s learn about metagenomics, including information on steps, applications, processes, workflow, analysis tools and techniques. Stay tuned.
Key Topics:
What is Metagenomics?
“Meta” means “huge” or “so many” and “genomics” means “entire genetic composition of an organism” which literally depicts that metagenomics is a study of huge or so many genomes. In the present context, it’s a study of the genomes of the entire microbiome or microbial community.
The term “metagenomic analysis” was first coined by Handlesman and Goodman et al. in 1988, marking the beginning of a new era in genetics. This approach involves investigating any environmental sample for the presence of nucleotide sequences. Each sequence represents and belongs to a particular microbe, microbial community, or strain and is determined.
This is a comprehensive concept of any metagenomic analysis. Now let’s see some of the historical events in the field of metagenomics.
| Year | Scientist(s) | Discovery |
| 1988 | Handlesman and Goodman et al. | Coined the term “metagenomics.” |
| 1991 | Pace et al. | Isolation and cloning of DNA from an environmental sample. |
| 1995 | Healy | Metagenomic isolation from Zoo Libraries. |
| 2002 | Breitbart et al. | The first time used shotgun sequencing and analysed 5000 different virus species from seawater. |
| 2003 | Venture C | Analyzed 2000 different microbial species from seawater. |
| 2005 | Schuster S | Published sequence data from an environmental sample. |
“Metagenomics is a direct genetic analysis of genomes present in an environmental sample”
Thomas et al. (2012).
Metagenomics is entirely opposite to conventional microbiology techniques, as we said. Instead of a tedious and lengthy culture-based approach, high-throughput sequencing determines the DNA present in any sample, readily.
Moreover, low abundance and uncultured microbial populations can be captured by nucleotide sequence analysis. Take look at the example
Suppose, a plant species grow faster and healthier in the soil near a water body while the same plant species remain retarded when growing away from the soil near the water body. There might be some sort of bacterial population or community in either water or nearby soil that triggers the growth.
Scientists collect samples from such sites and conduct metagenomic analyses. Lucrative and valuable information, we obtain are
- What type of microbial population is present?
- How diverse the entire community is.
- What type of bacteria, virus or other prokaryotes does the sample possess?
- Does it have a direct effect on plant growth and health?
- If so, what genes or genetic factors are involved?
- How it contributes to the ecosystem.
Data obtained from the metagenomic analysis can vary depending on the sample being studied. Interestingly, it can provide a wealth of data and information beyond what is currently known. To better understand it, we are discussing steps and a process in the present scheme.
Metagenomic workflow:
Technically the metagenomic workflow is complex. Here I have explained a typical and popular next-generation shotgun sequencing metagenomic analysis workflow.
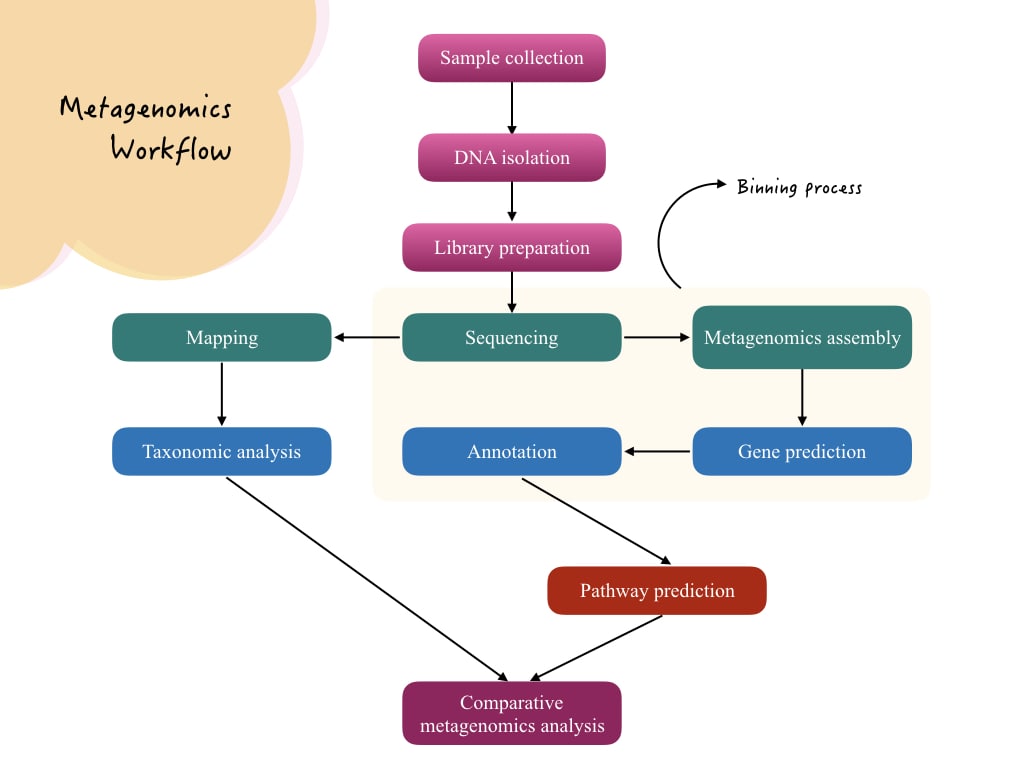
Definition:
Metagenomics is an interdisciplinary field of genetics that studies the entire microbial community present in the sample.
Process:
The process of metagenomic analysis is completed in the following steps. We will discuss each step one by one.
Sample collection:
The very beginning of the process is environmental or biological sample collection. Common sample types for analysis are soil, water (seawater, pond, river, dirty water), waste, and decomposing biological materials, feces and urine.
Potential biological samples are intestines, gut, stomach, oral cavity and human skin, etc. Notably, the sample doesn’t need any special treatment but will remain un-contaminated during collection, transportation and processing.
A strong recommendation for metatranscriptomics analysis, which is a study of gene expression, is a fresh sample collection and quick processing. Season, time, human interference and pollution are several factors having a direct effect on sample collection.
| Environmental samples | Biological samples |
| Soil, mud, water- pond, river, seawater, decomposed biological samples, | Stool, urine, animal- skin, intestine, gut, oral cavity, stomach etc. |
DNA extraction:
The entire success of metagenomic analysis highly depends on the DNA extraction– the choice of technique and how we perform it. Unlike other genomic extraction schemes, metagenomic DNA isolation is an entirely different and tedious process.
Ready-to-use kits for extraction are better options here for samples like soil, or any water but to obtain low-abundance DNA, magnetic bead beating is advised. In addition, for samples with a heterogeneous mixture of host cells, pre-processing is required to eliminate unwanted nucleic acid from sequencing.
To avoid genomic DNA isolation from the host cell, prior removal is necessary. Fractionation or selective lysis is performed to ensure minimal host DNA. Reviews discuss other techniques too.
Note that the protocol should be capable enough to produce high-yield and high-quality DNA. Quality and quantity assessment prior to sequencing is mandatory for getting better results. Poor-quality extract might be removed and the sample is reapplied for DNA isolation.
For samples with low-quantity DNA like any biopsy or water sample, enrichment is performed to improve the yield. Using random and known hexamer primers, DNA is amplified in a specialized PCR protocol.
PCR amplification isn’t required (to avoid primer or PCR bias) in the next-generation shotgun metagenomics sequencing platform. I have discussed the scheme of how NGS is applied in metagenomics. Click the link to read the article.
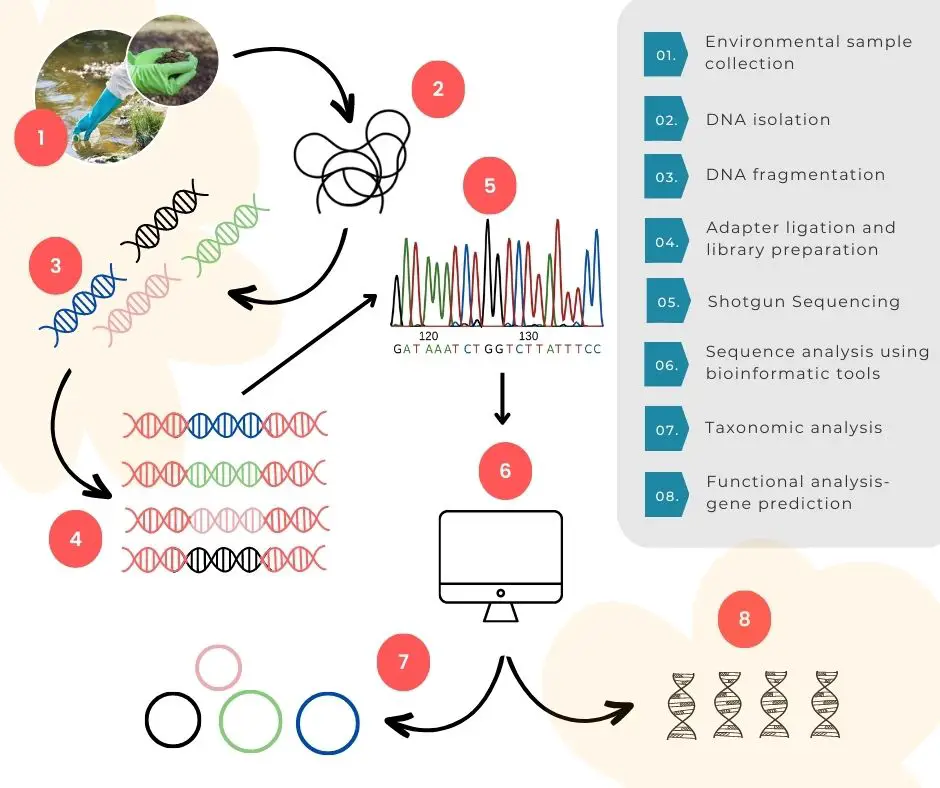
DNA sequencing:
DNA sequencing is a key step here, in the process. Sequencing reads the nucleotide sequences present in the sample. The old-school and conventional Sanger sequencing and various NGS platforms, both can be used.
However, NGS technology, like shotgun sequencing, is the most accurate, widely-used and common technique for metagenomics. Our already-published article contains all the necessary information on NGS. I recommend reading it.
Depending upon the platform, DNA is either fragmented into smaller pieces, cloned into a BAC vector or directly employed for sequencing. End result of any sequence would be a file containing nucleotide sequences– many, short & long and good and bad reads.
Note: In NGS sequencing reads are referred to as reading every nucleotide from the fragment that belongs to a particular library.
Common sequencing platforms used are enlisted here
- Traditional Sanger sequencing
- Pyrosequencing/ 454 Roche system
- Illumina/Solexa system
- SOLiD sequencing by applied biosystem
- Hiseq2000 by Illumina
- Single-molecule sequencing by PacBio
- Ion torrent and Ion Proton sequencing
The sequence read file is sent to assembly.
Assembly:
Assembly is a process to collect, arrange and re-construct reads to complete the sequence. Various computational tools are now available for different assembly processes and complete metagenomic analysis. De novo assembly and co-assembly are two common assembly options.
De novo assembly is performed when a set of reference sequences or reference genomes aren’t available to compare and arrange. Hence, the process is tedious, complex, time-consuming and requires immense computational power.
Co-assembly is a reference-based assembly process. Here, the target genome is compared with the reference genome or sequences available in any database. It’s a, comparatively, less-difficult process but needs high-end computer tools for assembly.
Nonetheless, sequence depth and coverage are two crucial factors that directly influence the assembly process and make it less valuable. This means that as the entire community of microbes, when assembled, contains different lengths, different numbers and different types of DNA sequences, it is hard to investigate the accuracy and quality of the assembly generated.
Moreover, unlike the high coverage genomic data, metagenomic sequence data are less- redundant and contain repeats too, which also makes the assembly process weaker. Now, after that, we have to estimate which genome belongs where! Here’s come binning.
Binning:
Thomas et al., 2012 define the binning process as grouping the assemblies to manifest closely related organisms. Sequence binning occurs in many ways (using computational tools) and how it can be done and what information it will give depends on the experience of the scientists.
For example, during binning, we get some unknown sequences, compare them with the already present gene database and come to know with which gene it’s closely related. Later, the function is determined and grouped in microbes that possess that particular function or sequence.
However, in the routine binning process, sequences are clustered by their relatedness to the reference genome. You can understand the binning process as showing which sequence belongs to which organism.
Gene Prediction:
Binning gives us a rough idea about what genomes or sequences we have and where they belong. For more rigorous analysis— for example, functional analysis, further computational processing is performed.
The next step is gene prediction. It allows prediction of potential genes present in the assemblies thereby protein produced by each if any. Eventually, it gives scientists a broad idea about the function of the entire metagenome.
This means, depending upon the presence of types of genes, what types and number of functions a particular community performs in the system, environment or ecosystem, can be determined.
If you think, it is enough; further processing is still performed.
Gene Annotation:
In the annotation, the predicted protein sequence and function are matched with the protein database available. This allows us to create relatedness based on functional analysis. Based on each protein and its function, and comparison with different protein families, functional classification is carried out.
How does it help?
It helps us to know how functionally diverse or related a microbial community present in the sample is. Interestingly, this is just one benefit of annotation, there are many lucrative applications this process has.
Nonetheless, the major limitation of annotation is that it could only be performed using the already existing annotated data, which is too low. A huge amount of data has still remained unannotated.
Advantages of metagenomics:
One of the uppermost advantages of the present technique is its power to study many microorganisms in a single experiment which could not be possible with conventional microbiology methods.
Besides, the sequencing-based metagenomic technique is highly accurate and faster.
We can also quantify the amount of viral load.
Disadvantages of metagenomics:
The techniques are so costly. Next-generation sequencing techniques cost around 500$ to 5000$ approximately.
So many microbes are still unknown to us therefore we don’t have enough information to compare and study novel microbe sequences.
Related article: Microbial genetics: A rapid advancement in microbiology.
Applications of metagenomics:
The metagenomics studies are now in their preliminary phase but it is the potential enough to penetrate different fields to solve different problems. It is used widely in ecology, environment conservation, infectious disease diagnosis, Environmental remediation, Biotechnology, and agriculture.
Biotechnology studies:
In recent times scientists have more focus on microbial studies viz metagenomic analysis. Protease, lipase, and nitrilases like enzymes are the product of metagenomic studies.
Enzymes, antibiotics, biochemicals, Bioactive compounds and pharmaceuticals are made by studying microbes only.
Ecological studies:
Microbial studies like metagenomics have great importance in ecology, conservation and invasive species studies.
Sea, rivers, soil, air, and rain forest are the habitat for so many different animals and microbes.
The complex symbiotic relationship between animals, microbes, and plants helps us to understand the health of the habitat. For example, the feces of one animal might be a nutrient-rich food source for another different species. Note that it might possible because of the microbes’ activity!
The metagenomic analysis provides insight into how both are important for an ecosystem.
It is also used for conservative and endangered species studies.
Healthcare and medical:
Complex infections can be immediately studied by metagenomic analysis.
Sample from the patient is taken and processed for DNA sequencing in order to know which microorganism may be present in them.
The role of different RNA viruses in human health can also be evaluated by isolating RNA and converting them into cDNA.
The impact of pollutants on the ecosystem and environment can be monitored and investigated by knowing how its microbial load behaves, by metagenomic analysis.
Agriculture and soil ecology
Metagenomics is tremendously used in soil and agriculture studies.
The soil is a common habitat for so many microorganisms and plants too. Approximately one gram of soil sample contains approximately 10,000000000 to 10,0000000000 microbial cells.
If we sequence all the microbes from one gram of soil sample, it gives 1 GB output sequencing information and that’s huge!
Complex relations between plants and microorganisms are the major focus of these studies. The microbiomes that are useful to plant growth have great economical value in terms of production.
Conclusively we can say, we can use metagenomics in different fields as per our vision. Even scientists are trying biofuel production through metagenomic.
Conclusion:
The process of metagenomic sample analysis might look familiar but it’s not only restricted to the sequencing of 16s rRNA gene analysis only. A lot of computational tools are used to get so much information related to the DNA sequences from a sample.
Thousands of different microbial strains can be identified, studied, and analyzed by the present technique. The world of microbes is even more complex than we think!
Sources:
National Research Council (US) Committee on Metagenomics: Challenges and Functional Applications. The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. Washington (DC): National Academies Press (US); 2007. 1, Why Metagenomics? Available from: https://www.ncbi.nlm.nih.gov/books/NBK54011/.
Thomas T, Gilbert J, Meyer F. Metagenomics – a guide from sampling to data analysis. Microb Inform Exp. 2012 Feb 9;2(1):3. doi: 10.1186/2042-5783-2-3. PMID: 22587947; PMCID: PMC3351745.