“To get success in the gel electrophoresis of DNA we need to use a proper concentration of each chemical, an exact concentration of agar, and need to do some routine optimizations.”
DNA is a helical structure in which two single-stranded DNA twist around each other and form a double-stranded stable DNA molecule. When we talk about a DNA, a structure like this comes to our mind,

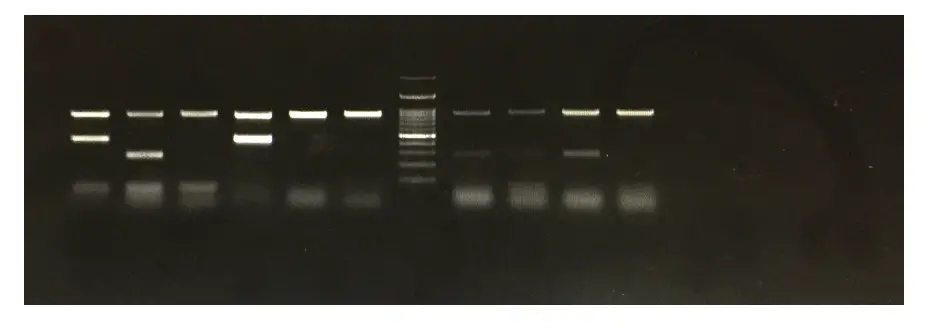
But can we observe or see the exact spiral structure of DNA like this? The answer is no. Not in routine experiments. Gel electrophoresis is a method to visualize or to detect DNA in a biological sample.
But a spiral DNA will not be seen in a gel. DNA in a gel looks like a thick and lightened band.
Two common gel electrophoresis methods are practiced to visualize DNA viz horizontal and vertical electrophoresis known as agarose gel electrophoresis and polyacrylamide gel electrophoresis, respectively.
However, the agarose gel electrophoresis method is easy to use in comparison with PAGE and henceforth routinely used in genetic labs.
Electrophoresis is a process in which the charged particles are separated based on their charge and size. We have covered an amazing article on the present topic. You can read it here: Agarose gel electrophoresis: Principle, Process, Type and Applications.
Practically speaking, we need an optimized protocol to get results in agarose gel electrophoresis of DNA. We should have to take care of some steps to get the best out of it.
Electrophoresis is practiced not only for DNA but also for other molecules like proteins. So we need some optimization and rule to follow in order to get results for DNA.
See, messy gel results are not explainable. DNA bands must be sharp, clear, and thick enough to analyze results.
In the present article, I will give you 10 of the proven tips to get success in the gel electrophoresis of DNA.
Related article: Agarose gel electrophoresis buffer.
Key Topics:
10 Proven Tips to Success in Gel Electrophoresis of DNA:
- The concentration of chemicals
- Choosing the right buffer
- A type of sample loading dye
- The role of glycerol
- The concentration of the agarose
- Say bye-bye to the smiley bands
- Loading the sample
- Quantity of the loading dye
- Re-use of buffer and gel
- Submerging the gel
The concentration of chemicals:
Consider a gel as a canvas to paint. You can picture anything in it. It depends on you, how you use different chemicals and this will decide how your picture looks like.
If you use a proper concentration of every chemical in the electrophoresis, your results get better and better. Trust me, The concentration of chemicals makes a difference in results. Use a concentration given in the charge below,
| Chemical | Concentration |
| Buffer | 1X |
| EtBr | 0.5microgram/ml |
| Agarose | As per the DNA fragment size |
| BPB dye | 1X |
The process of electrophoresis is a game of only 4 chemicals. But how we use it decides what we get.
Choose the right buffer:
To electrophoresis of DNA, we have two buffer options; TAE and TBE electrophoresis buffer. The composition of TAE is tris, acetate and EDTA and the composition of TBE is tris, borate and EDTA.
Both buffers can be used to run DNA of any size. But to separate larger DNA and smaller DNA fragments, We should choose a proper buffer system.
Go anywhere on the internet, no one will tell you which buffer to use when. From our personal experience, we can say,
To separate longer DNA fragments accurately, TAE buffer is recommended while to separate smaller DNA fragments TBE buffer is recommended by us.
To isolate DNA fragments of up to 1000 to 2000bp use TBE buffer and for fragments more than 3000pb use TAE buffer.
The concentration of different chemicals used in the buffer and its quantity are given in our previous article. Read it here: Agarose gel electrophoresis buffer.
A type of sample loading dye or tracking dye:
DNA can’t be seen by the naked eye. We have to track it by some color because it doesn’t have its own color or odor.
In the gel electrophoresis of DNA, we are using another special type of dye known as tracking dye to monitor the migration of DNA in a gel.
Bromophenol blue, Xylene cyanol FF and orange G are some common options for that which give pinkish-blue, sky and yellow color in a gel, respectively.
You can use any dye in PCR fragments or any DNA fragments though, BPB is highly recommended by experts.
But let me tell you that not all dyes are used in all types of DNA separation. The reason is the composition of each tracking dye is different and thus runs at different speeds.
For example, we are doing some restriction digestion and want to separate a fragment of 5000 bp, you can’t do it with orang G, why? It runs parallel to or above the 50bp fragment. And before the DNA fragment came out, the dye would run out of a gel.
It is worthless in this case.
A xylene cyanol, BPB and orange G can run parallel to 4000bp, 300bp and 50bp, respectively.
So ideally we can use BP for both smaller and larger fragments but to be precise use orang G for gel electrophoresis of smaller DNA fragments of up to 100 bp and Xylene cyanol for a larger one.
Bonus tips: don’t worry if you haven’t any of two, use BPB, it’s fine. The making of BPB is explained in our previous article. Read it Here: DNA Gel Loading Dye- Bromophenol Blue and Xylene Cyanol.
The role of glycerol:
You might be thinking that I had not mentioned glycerol and we are not using it in electrophoresis, right!
The role of glycerol is very very important to run a DNA gel. DNA can diffuse in a running buffer and comes out from wells. We need to sink it on the bottom of the gel so that it can pass through gel pores.
Glycerol gives density to DNA and makes it heavier than the buffer. By using glycerol, DNA remains at the bottom of the gel well.
But you don’t need to add it separately. It is already present in the loading dye. With BPB or orange G, the glycerol is added, already. Usually, 2.5 to 3% glycerol is added.
Related article: 10 Secret Tips for DNA Extraction to Get Good Results.
The concentration of agarose:
One of the most important factors in gel electrophoresis of DNA is the concentration of agarose powder. If you master this skill (when to use which concentration of agarose), trust me you will always obtain great results.
For example, to run a gel of gDNA or genomic DNA, use 0.8% agarose gel. It will separate gDNA properly, If you use 1 or 1.5% concentration, your gDNA bands got sheared.
Likewise, use 1.5% agarose gel to separate PCR products. The concentration of agarose and DNA fragment size is given in the table below,
Concentration of Agarose | Types of DNA sample | approx. fragment size |
0.8% | Genomic DNA | > 1 kb |
1.0% | PCR product and plasmid DNA | 400bp – 10kb |
2.0% | PCR product | 50bp – 2kb |
3.0% | Restriction digestion | 10bp to 1000bp |
Say ‘bye-bye’ to smiley bands:
You might come across a smiley band-like effect during gel running. What will it cause? Obviously, DNA bands can’t separate properly and we can’t estimate their size.
There are two reasons for that, improper gel concentration and high electrical current.
If you run a gel on high voltage DNA trying to run fast but due to the small pore size, it can’t! Consequently, you will get uneven, smiley bands.
Run gel on a voltage between 80 to 150V. For larger DNA fragments I strongly recommend reducing the input voltage to 80 or 60V. In other cases, if you have not prepared a gel properly and evenly you may see an uneven banding pattern.
Loading the sample:
You should learn sample loading because it is a difficult thing. At first, everyone breaks gel while loading a DNA sample.
I don’t know how to explain it to you. But I can say, load a DNA sample on the bottom of the well but do not break the well otherwise, your DNA can’t run in a gel.
You have to load the sample carefully. Once you become an expert in it, you can do it easily. So practice hard to load DNA samples properly.
Quantity of a loading DNA:
You can’t add 100microliter of sample to a well, right! Because it will come out and mix with other DNA samples.
To avoid it, you must have to insert an amount of sample which can’t leak from a well. I advise adding 7μl of DNA and 3 or 5 μl of dye, mixing it on a parafilm and load into a gel.
Re-use of buffer and gel:
If you are planning to reuse a buffer or a gel, it’s fine. We can do it. But repeated use of buffer and gel can retardate our DNA gel results.
Every time we heat the agar its concentration changes (reduces). But we can use it twice or sometimes thrice but not more than that. In addition to this, to re-use a gel, run all the DNA fragments out of the gel before reuse.
You can re-use the running buffer twice or thrice as well but as you re-use it more, you will notice abnormal gel results. It might be shearing, smiley bands, no separation, improper migration of DNA, anything!
However, to use DNA gel electrophoresis in medical or disease diagnosis, don’t reuse things. But for demonstration purposes for some research, you can re-use it.
Submerge the gel completely:
If you don’t fill the gel tank completely until the gel is submerged in it, your sample can’t run. Your gel will melt or the DNA band will not migrate properly. Fill the gel tank completed above 1 to 1.5cm of the gel surface so that gel can submerge in it properly.
Related article: A complete guide for analyzing and interpreting gel electrophoresis results.
Conclusion:
For the gel electrophoresis of DNA, we need agarose, gel electrophoresis buffer, loading dye and the electrophoresis unit. We have performed gel electrophoresis thousands of times, trust me these tips will surely help you in your experiments.
Furthermore, the quality of chemicals used in it also plays a crucial role, hence choosing the manufacturer carefully. A good quality agar can solve many problems.