“Determining the nucleotide sequence orders of the whole exome set of an organism using a high throughput DNA sequencing method is known as whole-exome sequencing.”
Three different biomolecules construct a DNA viz pentose sugar, nitrogen bases and triphosphate. All DNA is arranged on chromosomes which makes a genome of an organism. All the somatic cells have the same genome or genetic content.
The genome comprises coding DNA and non-coding portions of 3% and 97%, respectively. The coding portion is known as genes that make various proteins. Genes are made up of a sequence of exons as well as introns and lead by promoter region.
Here the introns are non-coding intervening sequences that are removed during the mRNA translation. Only exons remain there to make a final product- a protein.
The technique used to sequence only the coding exons is known as exome sequencing. But why only exons are being sequenced? And what is the exome?
In the present article, we will try to explain what an exome is and how the whole-exome sequencing platform works.
Key Topics:
What is an exome?
As we said, only the exons of a gene are capable of manufacturing proteins. And the intervening sequences are removed during the transcription and thus do not take part in final mRNA formation.
A gene is made up of not only a single exon but also there are one or more than one exons in a gene. The entire set of all the exons of a genome is known as an ‘exome’ or ‘whole exome’. What does it mean?
Simply put, the entire set of only a coding region of all the genes of an organism’s genome is known as an ‘exome’ or ‘exome set’ or ‘whole exome’. The exome comprises only a 2% portion of the entire genome.
Majorly, mutations occur in the coding regions of genes are more harmful. Thus, If we sequence the entire set of exomes we can get information on the mutated exon, a genetic disease associated with it and its phenotypic effect.
Information: a total of 180,000 exons are present in the human genome which consists of 1 to 2% of the total genome.
Related article: What Is DNA Sequencing? A Beginners Guide.
What is whole-exome sequencing?
DNA Sequencing is performed in order to get sequence information. A sequencing machine identifies and detects every nucleotide present in a target sequence or in a gene.
The whole-exome sequencing is a next-generation high throughput DNA sequencing technique powerful enough to sequence the entire coding region of a genome.
The first sequencing method was developed by Sanger and known as the Sanger sequencing method, which is still used in laboratories to sequence smaller DNA fragments of up to 1000 to 1500 nucleotides.
However, the Sanger method is not so fast thus to sequence the entire genome; it takes several years! On the other side, the next-generation sequencing method is so fast that it can sequence the entire genome of 3 billion bases within a day to a week.
The whole-exome sequencing method can sequence the entire exome set. Hence more than 85% of disease-causing variants can be sequenced or detected.
Mutation in coding regions is considered more damaging, however, several mutations outside the coding region can also affect one’s health adversely. For instance, the IVS 1-5 and IVS1-1 mutation of beta-thalassemia.
But as we said more than 80% of genetic disease-associated mutations, alterations and copy number variations are in the coding regions, means, in the exomes.
Sequencing a set of exomes is more feasible instead of sequencing the whole genome. Whole-genome sequencing takes more time and also is a costlier process.
Conclusively, the technique of whole-exome sequencing evolved to overcome two limitations viz to find out various variants, allele, or loci associated with a disease in cost-effective assays. Notably, it sequences not only protein-coding regions but also exons of other microRNA or lincRNAs.
Process of Whole Exome sequencing:
The process of this sequencing platform is a bit similar to the next-generation sequencing or other high-throughput sequencing methods.
The process is also somehow complicated, but here I am trying to explain it to you.
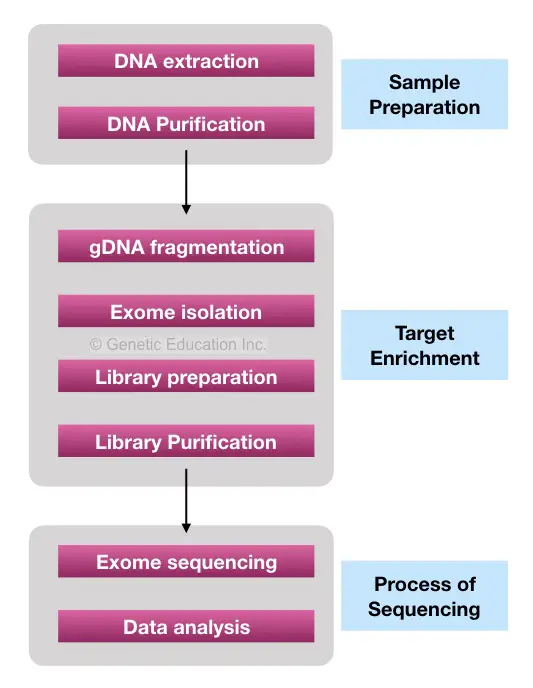
First, high-quality DNA is isolated using the salt-out method or ready to use the DNA extraction method. However, I strongly advise a DNA extraction method recommended by the manufacturer of the Exome-sequencing.
Two criteria should be fulfilled by any DNA extraction methods to use it in whole-exome sequencing, one the quality of DNA should be nearly 1.80 and the quantity above 100-250 ng.
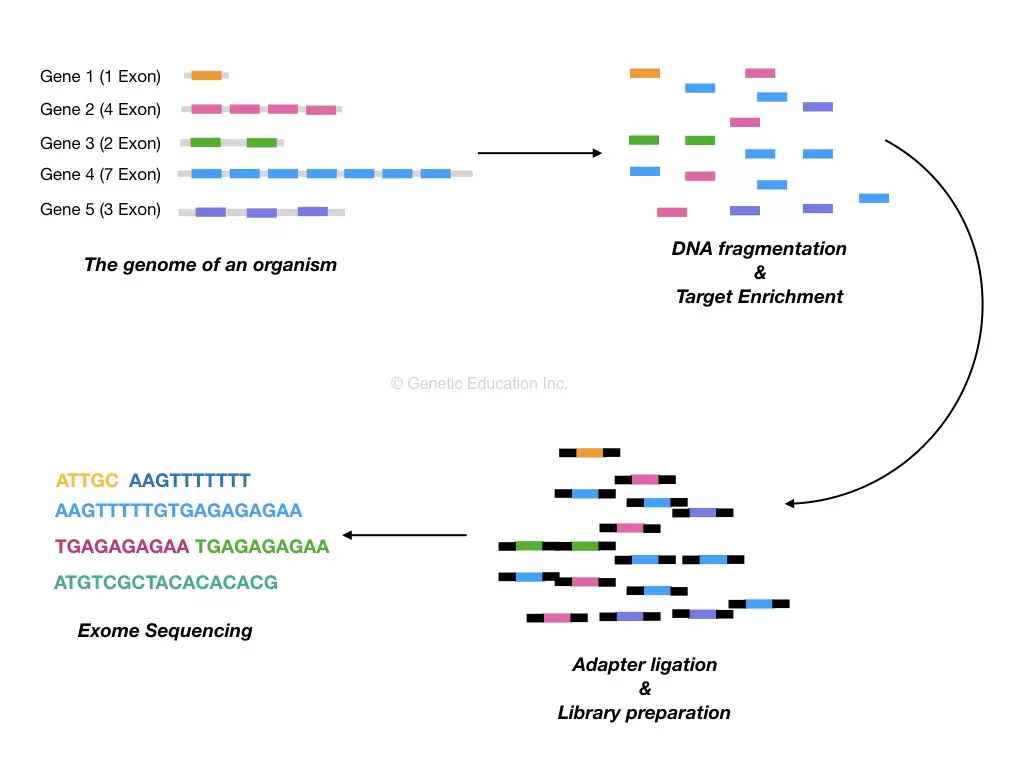
DNA extraction is followed by a target-enrichment process. In a target-enrichment process, the entire exome set is isolated from a DNA sample. Physical or enzymatic methods are performed in order to fragment DNA before making target enrichment.
Now come to the DNA fragmentation,
The intention to make DNA fragments is to decrease the complexity of the assay. Larger DNA fragments are hard to sequence henceforth we are making small fragments of DNA and collecting it in a library. A library is a known place where all our DNA fragments are located.
The fragmented DNA is processed to library preparation in which blunt ends are generated and ligated with the adapters.
Target Enrichment:
Target enrichment is nothing but the methods or collection of methods used to select the genomic regions we wish to study or sequence. In our case, the “exomes!”
Using methods like the Array-based capture or In-solution capture method, we can select and isolate the regions we wish to sequence, such as Exomes.
The array-based capture method is one of the traditional methods in which the probe specific to our target DNA sequences are immobilized on the solid surface.
The exome sequences are hybridized on each probe (having its complementary sequence) and then amplified in the PCR.
However, steps like producing blunt ends, adapter ligations and washing are also included in it like the conventional microarray. The requirement of prior sequencing information, tedious sample processing makes it hard to use.
In the solution capture method, the probes are there in the solution (of beads or magnetic beads) on which the fragment of DNA is hybridized. The rest of the unhybridized DNA is washed off and the beads with the DNA fragments of our interest are sequenced.
So these are the two methods to make target enrichment for sequencing the exomes.
Once the sample preparation is completed, the washing step is performed followed by library purification. We fragmented DNA thus we have our exons fragments as well other DNA fragments that we do not wish to sequence. Washing removes all the other non-required DNAs along with traces of other chemicals. By eluting the sample a pure library of exon fragments is ready for DNA sequencing.
The library is processing for sequencing. The sequencing machine reads each sequence of the entire exome library and gives an output in the form of signals.
To obtain millions of short-sequence specific reads massively parallel high throughput sequencing is performed.
We have covered a dedicated portion in which we have explained how massive parallel sequencing is done. You can read it here: Next-generation sequencing.
In short, in a bridge-amplification process, every ddNPTs after every elongation stage is detected in an NGS that is the whole mechanism. The computer software arranges every nucleotide orderly and determines the sequence of every fragment.
Based on the library arrangements, individual fragments are arranged and the entire sequence of exome is generated by a computer.
Related article: Preparing “DNA for Sequencing”.
Advantages and applications:
The present sequencing method is high-throughput, fast, reliable, comparatively cost-effective and accurate.
It covers the entire coding region of the genome.
The sequencing data is only 4 to 5 Gb output in comparison to 90 to 100gb of whole-genome sequencing.
It is used to known disease-related genotypes and alleles.
To find our alteration in coding regions associated with an abnormality.
It is used in screening genetic disease, identification of new variations and mutations, population genetics, cancer genetics and other fields.
The present method is highly accurate and therefore used so often for the multigenic disease. For example, genes mutations of Kabuki disease, paroxysmal kinesigenic dyskinesia, congenital chloride diarrhea and spinocerebellar ataxia are screened by whole-exome sequencing.
It is also applicable in discovering the new Mendelian disorder associated variants and identification of rare variants and gene mapping.
Limitation:
Exome-sequencing is a state of the art technique to sequence the DNA, we know! but it has one crucial limitation. Not all the disease-causing mutations are located on exons. Even though a broad spectrum of mutation associated with a disease or group of the disease can be encountered but some remains left.
To cover mutations, alterations, or copy number variations of the whole genome, we need a whole-genome sequencing method like shotgun sequencing or clone by clone sequencing.
And that is not possible in this method.
Yet another limitation of the present method is the sequence similarity of some exons. Not all exons are the same but some are not different too! Several exons have repetitive DNA sequences shared between other exons. Those can’t be predicted correctly.
Conclusion:
The whole-exome sequencing method is faster, cheaper in comparison with WGS and accurate. It can give sequence information of not only mRNAs but also exomes of siRNA and miRNA.
Nonetheless, extensive computational knowledge, high-speed supercomputers, and expert manpower are necessitated in order to get results.
In addition to this, laborious electrophoresis steps are not needed in it. Exome sequencing is beneficial in clinical, research, and academics. Prognosis, diagnosis, and pathophysiology of a disease can be analyzed as well.
The present method is widely used to study non-mendelian or rare Mendelian disorders and to find our rare genetic variants that cause disease. Variants present in a small number of populations can be detected as well.