
“Sickle cell anaemia is an autosomal recessive form of an inherited condition, occurs due to the mutation in an HBB gene results in sickle shape RBCs and severe anaemia.”
Often known as SCD- sickle cell disease, SCA- sickle cell anaemia, HbS disease or haemoglobin S deficiency it is a type of rare haemoglobinopathy.
It was discovered in early, 1900 though, Dr Linus Pauling who was the pioneer in sickle cell research suggested that an abnormal form of the haemoglobin, known as “Hemoblogin S” is the reason for the sickle-shaped erythrocytes. He had done this research in 1951.
Further, he was stated that vessels blockage due to the sickle shape of the RBC is the major problem associated with it. Globally, the prevalence of the disease is 1 in 500 to 1 in 1500 in high-risk populations like African-Americans.
The red blood cells are the major target site of the present condition. The oxygen-carrying haemoglobin located on it involved in the SCD.
The abnormal beta-chain of the haemoglobin results in SCD. Vessel blockage, severe pain and anaemia are observed commonly.
In the present article, we will explain to you all about the sickle cell anaemia and related complications. We will also discuss its genetics and mutation associated with it. Furthermore, I will give you a standardised protocol which you can use it for either sickle cell anaemia or sickle cell carrier screening.
- What is sickle cell disease?
- Definition
- Causes
- Symptoms
- prevalence
- Genetics of sickle cell anaemia
- Mutations
- Sickle cell trait
- Diagnosis
- Treatment
- Prenatal genetic screening
- Managements
- Diagnosis of SCD using PCR
- My experience
- Conclusion
Key Topics:
What is sickle cell disease?
A sickle cell disease or sickle cell anaemia affects mainly the haemoglobin of the RBC. Because of the different variants of the haemoglobin chain, more precisely we can say it is a group of conditions rather than a single disorder.
It mainly affects the blood system, heart, lung, brain and liver.
Definition:
“An autosomal recessive form of the condition, sickle cell anaemia is a blood disorder occurs due to the mutation in the HBB gene results in anaemia, severe pain and blood vessel blockage.”
Symptoms:
Some of the common sign and symptoms of SCD are enlisted here, however, the symptoms vary from person to person.
Blockage of blood vessels,
Due to the abnormal haemoglobin, the rounded- RBC becomes sickle-shaped and hence blocks the blood flow. Chest, abdomen, joint and heart are the areas where the blockage is commonly observed.
A person suffers from severe pain because of this reason. This may last up to a day or even months.
Notably, the condition is known as a “pain crisis” or “sickle cell crisis”, which is indeed one of the common and dangerous symptoms of the present condition. Even normal painkillers can’t control pain crisis.
Vital organs of our body can’t function properly because of decreased oxygen-carrying capacity of haemoglobin.
Sometimes it causes acute chest syndrome in which blockage of the blood vessel is another reason besides the abnormal haemoglobin.
Higher blood pressure is more commonly observed due to the same reason.
One of the common and most dangerous symptoms of the SCD is the frequent bacterial infection. As we stated, the lung function decreases greatly, bacterial infections frequently happens.
The number of red blood cell also decreases- hence results in anaemia.
As the sickle cell shaped RBCs aren’t common, the abnormal cells undergo apoptosis more rapidly.
Conclusively the symptoms of the present condition are:
- Sickle cell crisis/ pain crisis
- Anaemia and fatigue
- Frequent bacterial infections
- Heart, brain and lung injuries
- Acute chest syndrome
- Stroke
- Pulmonary hypertension
- Difficulty in breathing (sometimes)
- Swelling in hands and feets
Causes:
The haemoglobin is the major site affected in the present condition.
Among all blood cells, only the RBCs- red blood cells have the oxygen-carrying capacity. Oxygen is badly needed for each and every organ of our body. Haemoglobin- a type of protein is a vehicle transfer oxygen molecules to every bodily organ.
Henceforth it is crucial for our survival. Structurally, the haemoglobin is made up of two parts- “haem” and “globin”.
The haem part, made up of four iron atoms are surrounded by four different globin proteins. A tetrahedral globin has two alpha chain and two beta chain. Both proteins are encoded by two different genes HBA and HBB.
Mutation in the HBB gene causes abnormal beta-globin chain which referred to as haemoglobin S or HbS.
A single base change in the HBB gene causes an abnormal amino acid chain in the final beta-globin chain.
Unlike the regular HbA (or haemoglobin A), the HbS chain is different, if both the globin chains become mutated, the cell becomes sickle-shaped and causes sickle cell disease.
The sickle-shaped cells are harder and less flexible compared to round shaped RBCs.
Genetics of sickle cell anaemia:
The HBB gene encodes the beta-globin chain of the haemoglobin. It is located on the p arm of chromosome 11. Cytological location of the HBB gene: 11p15.4 (between 5, 225, 464 to 5, 227, 071 basepairs)
Due to the point mutation in the beta-globin gene, a single nucleotide A converts into T. The conversion leads to change in an amino acid glutamic acid to valine in the beta-globin chain.
The new allele or variant is known as HbS, as we said earlier.
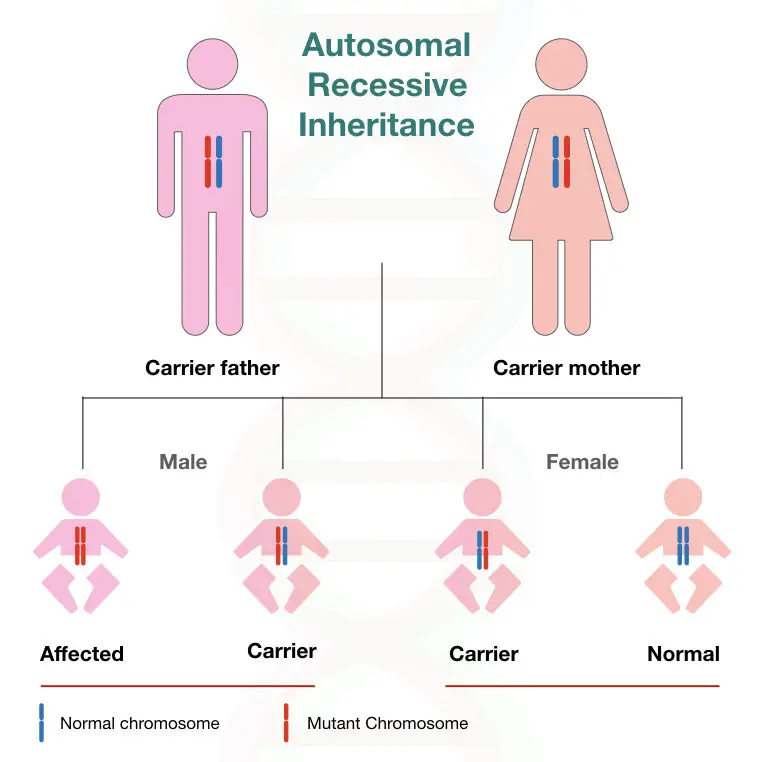
Interestingly, a single HbS allele is unable to cause sickle cell, if two HbS/HbS alleles inherited, abnormal haemoglobin is formed. The rest of the story, as we said earlier happens.
Sickle cell anaemia is an autosomal recessive condition. The disease symptoms appear only when two recessive alleles inherited into progeny.
Notably, an individual with only a single HbS allele remains normal, throughout their life. Any symptoms of the disease are not at all observed in them.
Interestingly, in some other cases, along with one abnormal beta-globin variant, another different allele of it occurs, for example, HbSBetathal. In this case, along with the HbS, another variant or allele for the beta-thalassemia occurs in an individual.
Sickle cell trait:
Now we know that a person with a single HbS allele is absolutely fine. A person with a single HbS with normal HbB alleles is called a carrier or more precisely “sickle cell trait.” A disease gene inherited from them to their offspring.
Interestingly, the sickle cell trait was evolved for a novel cause. It’s actually helpful for us.
Is sickle cell helpful?
Varieties of traits are evolved to make up survive on earth. Different alleles are originated for this cause, so the sickle cell trait too.
Actually, a carrier of sickle cell anaemia remains normal throughout life and it facilitates an outstanding survival cause.
It provides protection against a well-known parasite plasmodium falciparum– “malaria.”
As we stated above, the sickle cell trait is most prevalent in the African people or African subcontinent where the malaria is seen commonly.
The P. falciparum can’t identify the sickle-shaped RBCs and hence can’t infect. The Hbs trait is evolved to make those people survive against malaria. Isn’t it fascinating!
Conclusively, we can say, the SCD is not harmful or lethal, indeed it was one of the evolutionary factors that actually helped humans to survive.
Some literature suggests that a person with sickle cell trait may have some lesser severe complications. Those are not lethal, though.
Life expectancy:
Unlike other complicated, inherited genetic conditions such as trisomy 18 or 13, the mortality rate of the present condition is very less. Due to the recent advancement in genetic diagnosis and prenatal screening, a person with the sickle cell can live up to 40 to 60 year.
Although, extensive care and treatment needed.
Typically, a person with the sickle cell anaemia don’t die early but in severe conditions, the sickle cell crisis or pain crisis is unbearable. Even, routine pain killers can’t help in it.
However, a carrier or sickle cell trait people live a normal and healthy life without any complications.
Severe pain attack can happen in adverse conditions like stress, high temperature or dehydration.
Management & Treatments:
Varieties of treatment options are now available for sickle cell anaemia, thanks to recent scientific advancements!
People with SCD can surely live a long and healthy life. But until now, the patient could not survive up to even adulthood.
Let’s discuss some of the best treatments used for sickle cell disease.
Blood transfusion:
Blood transfusion is a process using which blood injected into the patient’s body artificially.
Blood transfusion working effectively for sickle cell disease. As we mentioned, blood vessel blockage is the prime reason for stoke and pain in the present condition.
Through blood transfusion, fresh flow of normal blood (RBCs) injected into the patient’s body.
The defective RBCs are replaced by a normal and healthy one and the patient may feel relief. Some complications are also associated with it, however.
Due to excessive blood transfusion, the amount of iron dramatically increases in the patient’s blood. This can cause serious health complications.
Chelation therapy:
Excessive iron in patient’s body increases the risk of cancer, infection, diabetes and other health complications.
Iron chelators are given simultaneously along with the blood transfusion. Chelation therapy is an important part of sickle cell anaemia treatments.
Antibiotics and vaccines:
The individual with the SCD is more prone to bacterial infections. Lung functioning reduced greatly by this therefore routine antibiotic such as penicillin is given very early.
Proper and on time vaccination are mandatory too for sickle cell anaemia.
Pain relief:
Hyddroxyurea, often known as hydroxycaramide is a type an anticancer drug, previously used in the chemo.
However, it is adopted recently as a treatment option for curing sickle cell crisis.
The drug stimulates the production of foetal haemoglobin, HbF- another form of the normal haemoglobin. This HbF reduces the HbS, and because the foetal haemoglobin is not affected by the mutation, it can transport oxygen effectively. Nonetheless, the effect of the drug is not prolonged.
Bone marrow transplantation:
All the blood cells are originated and differentiated from the bone marrow. Hence if we replace the patient’s bone marrow with someone’s healthy marrow, the sickle cell disease can be prevented.
The bone marrow transplantation is the only option available for curing the SCD, however, the success rate is only around 18 to 20%.
In this treatment, the faulty bone marrow of the patient is replaced by his or her genetically compatible relatives.
Here, it is very important that the donor must be genetically related or compatible with the receiver.
Once it is implanted, new RBC cells without the mutant HbS allele are divided from the healthy bone marrow. But unfortunately, the success rate is not much. Besides, other complications are also involved in it.
Apart from all these, by adopting a healthy lifestyle, the patient can overcome complications. For instance, by drinking plenty of fluid risk of sickle cell crisis reduces, also, by avoiding unhealthy and contaminated environment chance of infections can be decreased.
Moreover, regular medical check-up and doctor’s follow up is required to avoid problems related to sickle cell disease.
Gene therapy:
What if we replace the faulty HbS allele with a wild type HBB!
Due to the tremendous progress in genetic engineering technologies, sickle cell anaemia can prevent using gene therapy.
First, let’s understand some basics of gene therapy.
A faulty, mutated or abnormal DNA sequence or a part of the gene or an entire gene is replaced by the healthy- wild type DNA- the process is known as gene therapy.
In gene therapy, either DNA is inserted or deleted. In Sickle cell anaemia, the faulty HbS allele can be replaced by the normal one using either viral or non-viral vectors.
Practically, the entire process is practised in vitro. Healthy cells with the new gene are cultured using the tissue culture methods and inserted into the bone marrow cells.
However, gene transfer techniques are now not ready for routine clinical practises because of the complications associated with it.
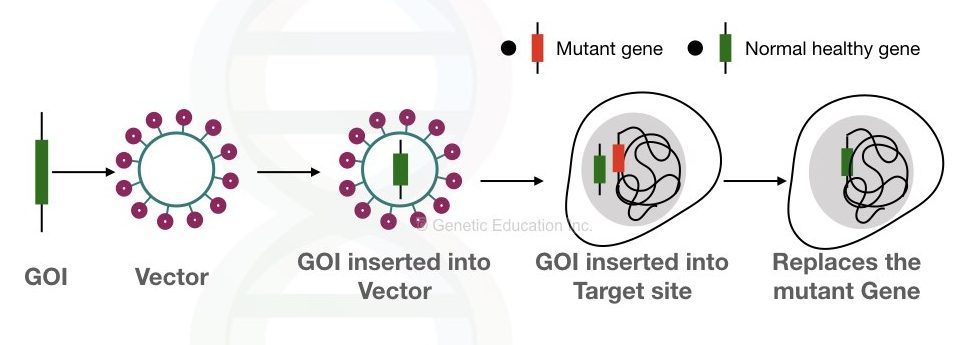
Despite having a low success rate, gene therapy is a promising approach helps in future for curing sickle cell anaemia. Besides this, stem cell therapy is also an option for curing SCD.
Read more:
- Gene Therapy: Types, Vectors [Viral and Non-Viral], Process, Applications and Limitations
- What is Gene Therapy? and How Does it Work?
Screening and Diagnosis:
As the disease is actually not lethal to us, there are several really outstanding diagnosis method for postnatal and prenatal diagnosis are available nowadays.
The primitive test used for the sickle cell screening is routine microscopy. The normal RBCs are doughnut-shaped while in SCD it becomes crescent moon-like or sickle-shaped.
By observing the shape of RBCs, the screening was performed, although the carrier can not be distinguished by the present method.
Another method for screening the present inherited genetic condition is the sickling method.
Using the specific buffer, I named it as “sickle cell buffer”, the sickle-shaped cells can be precipitates. Based on the naked-eye observation, sickle cell disease can be screened.
Still, here also, carrier detection is not possible.
DNA testing:
The DNA testing method is the only trusted and accurate method used for the diagnosis of SCA. Furthermore, a great advantage of the present method is that a carrier can be detected by the HbS-DNA analysis.
The HbS-DNA testing is advantageous for prenatal as well as the postnatal finding of sickle cell anaemia.
The DNA testing method is 99.10% accurate, reproducible, rapid and trusted.
Here, first, a patient’s DNA is extracted- purified and quantified (for routine lab quality control). A set of the mutation-specific and normal allele-specific primers are employed for amplification in PCR.
If the mutant alleles amplify, the sample is sickle cell positive.
If mutant and normal both allele amplifies, the sample is a carrier.
If only normal alleles amplify, the sample is normal.
The PCR based sickle cell testing is one of the simplest method of DNA testing, no fancy PCR optimization needed.
However, internal positive, as well as a negative control, must require to conclude the results.
If we go one step ahead of the PCR testing, DNA sequencing can also be performed. The HBB gene sequenced for knowing the mutation. However, in most cases, almost a single mutation is present in HbS allele.
Parental genetic testing:
Prenatal genetic testing for sickle cell anaemia is a powerful weapon for the prevention of it.
The method is the same, as detailed above, however, the sample collection is a crucial catch here.
Through amniocentesis, a sample is collected, also chorionic villi sample also preferred in an early stage of pregnancy. Thus the method is a bit risky due to the chance of miscarriage in it.
Moreover, expert hands needed for extracting DNA from either amniotic fluid or chorionic villi because extracting DNA from a fetus sample is not an easy task.
Notably, using the cell-free DNA present in maternal blood, non-invasive screening is possible. We have discussed cell-free DNA testing in our previous articles. You can read it there.
Evidently, the major advantage of prenatal genetic testing is that one can know about the fate of the fetus in terms of sickle cell anaemia.
Conclusion:
Sickle cell anaemia not lethal still, preventive action must require due to the dangerous health issues associated in it. Pregnancy genetic testing is the best option for preventing it.
If any of your family members is a carrier for the sickle cell disease, you must have to go for DNA testing.

Great content! Super high-quality! Keep it up! 🙂