“Transcriptomics is a study of transcriptomes- entire RNA transcript present in a cell or cell population to investigate gene expressions.”
In the present article, our major focus will be on first “omics” and second, “transcript”, which is obvious. But before that, we have to understand several basics that make a platform for understanding the topics well.
The field of transcriptomics evolved in the ‘90s with promising approaches to study gene expression.
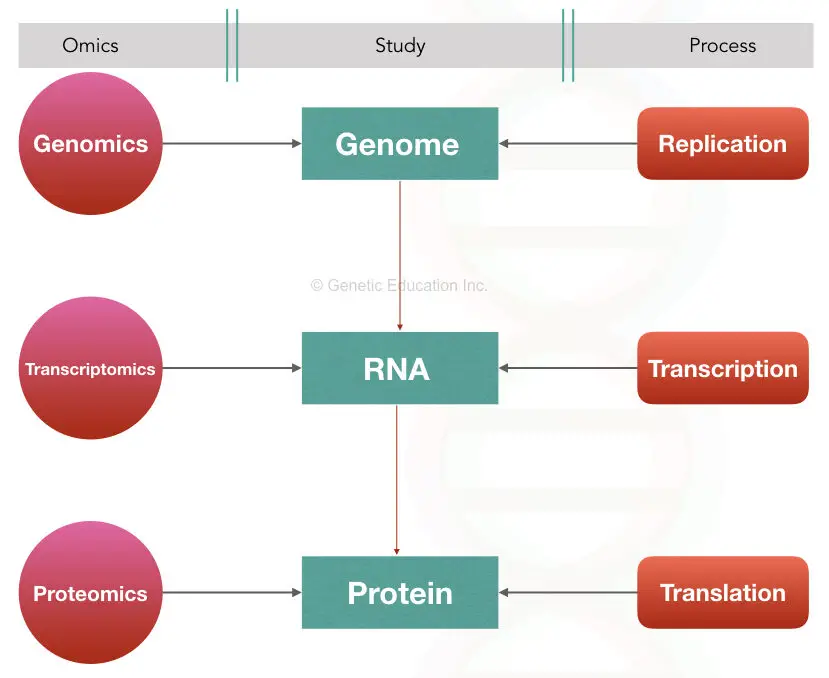
The journey of DNA to protein through mRNA is called gene expression.
Or we can say when a gene creates a protein, it is expressed, otherwise not. Therefore it is obvious that various genes expressed differently in different cells, different tissues and under different conditions.
For example, under the high sunlight, a gene for melanin expressed higher in skin cells rather than lung cells.
Consequently, it is possible that the gene expressed abnormally. Thus an abnormal mRNA- a transcript, and abnormal protein can also form. These are studied in genomics, transcriptomics and proteomics, respectively.
Key Topics:
What is omics?
Genomics: The study of genes or DNA or genome is genomics
Transcriptomics: the study of the mRNA or transcript and related technologies are called transcriptomics
Proteomics: the study of protein, related alterations and technologies are called proteomics.
Transcriptomics, an omics that is associated with the gene expression and mRNA or we can say RNA studies. In the present article, we will discuss the transcriptomics and related technologies.
The content of the article is,
- What is a transcript?
- Definition
- Study
- What is transcriptomics?
- Definition
- Process
- Transcriptome data analysis
- Single-cell transcriptomics
- Applications of transcriptomics
- Conclusion
So let’s start the topic,
What is a transcript?
A gene is a coding entity of the genome which has the power to form a protein. First, it is transcripted into the mRNA in which the non-coding DNA sequences are removed; now this mRNA is our transcript.
The mRNA migrates from the nucleus to the cytoplasm- at the ribosome and by taking the help of tRNA and rRNA, a protein is built from the mRNA.
Thus, not the entire gene but the transcript of it- mRNA can translate into an amino acid chain.
Definition:
“The transcribed mRNA of a particular gene is called a transcript.” Or
A transcript is an mRNA related to the particular gene.
What is transcriptomics?
Definition:
“The branch of genetics, that study the total RNA or mRNA present in a cell or tissue and related technologies used to study it, is called transcriptomics.”
Or
“Using the high throughput technologies such as microarray or RNA-seq; studying the transcriptome- a total set of mRNA present in a cell is known as transcriptomics.”
In 2008, the full transcript of 16,000 genes was published successfully, however, the first transcriptome of 609 mRNA is published during 1991.
Transcriptome-
Depending upon the type of experiment, a total set of mRNA (or sometimes all RNA) present in a sample is called transcriptome.
By studying the transcriptomes we are studying not only the gene expression, but it also includes the study of all the forms of RNA.
Abnormal tRNA or rRNA can also cause abnormal gene expression hence it is also included in the transcriptomics study. In addition to this, non-coding RNAs such as microRNA or siRNA are also covered in it.
Interesting, here we not studying mutations or sequence variations, because we are dealing with the final product- a protein.
95% of the mRNA can not form protein, henceforth, by analysing the transcriptomes we can determine which protein is produced or which is not.
Eventually, in a particular cell, the amount of active gene expression can be estimated.
This is the whole idea of studying transcriptomics. However, a vast amount of data is required to compare the results. Technologies such as microarray, RNA sequencing and reverse transcription PCR is used to do so.
Although, for entire transcriptome study either microarray or RNA seq is more preferred.
The transcriptome is the all set of RNA- mRNA, tRNA, rRNA and other non-coding RNA presents in a cell.
Process of transcriptomics analysis:
RNA extraction:
The process of transcriptomics starts with the extraction of RNA. Depending upon the experimental requirement either total RNA or mRNA is extracted from the tissues.
However, the process of extracting RNA is further a tedious job to do hence expert hands must require for transcriptomics study.
Extracting RNA is a challenging task because it is weaker than DNA and thus can be fragmented easily either by external conditions or by the RNase.
Also, sterile conditions must be strictly maintained during RNA extraction, because RNase is present everywhere- on our hands, on a desk or even on a pipette.
Ready to use RNA extraction kits are strongly recommended for doing this, although those are still not well standardised.
All we can say is, extracting RNA is an art- the purity of the extract depends on the experience of the researcher.
Anyway, now our pure mRNA is ready for downstream processing.
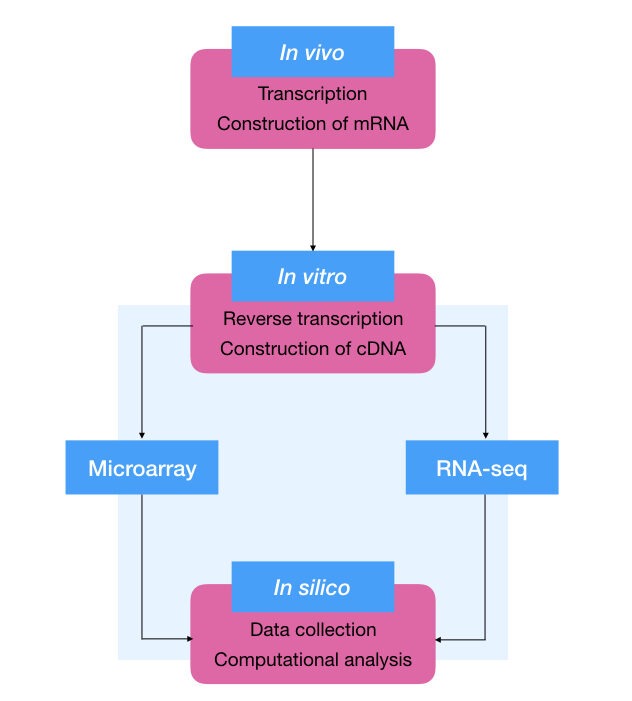
Reverse transcription PCR:
Using the reverse transcription PCR method, a cDNA is constructed from the mRNA.
A special type of polymerase called reverse transcriptase-polymerase, construct DNA from the mRNA.
(Transcription: from DNA to mRNA, reverse transcription: from mRNA to DNA).
Now the cDNA or complementary DNA is ready.
For more detail on protocol and cycling condition for reverse transcription PCR, read our previous article: Reverse transcription PCR.
The cDNA is ready for the next step of transcriptomics analysis. It is processed either through the microarray or RNA- sequencing.
Transcriptomics analysis:
Microarray and RNA- seq are two methods used for transcriptomics analysis. Besides this, RT- PCR is also used for gene expression studies.
Microarray:
The microarray is a special type of set up in which the probes are immobilised on the solid glass surface.
The probes are designed using the previous data or sequence information from the sequence tag site database. Read more on how to construct a probe: DNA Probes: Labelling, Types And Uses.
The sequence tag library is a collection of the short sequence of expressed genes of DNA.
Using this data, probes are constructed and immobilised on a solid surface.
The sample (which is our cDNA reverse transcribed from the mRNA) is fragmented using restriction digestion method and labelled with the fluorochrome.
The sample, as well as the control- DNA or cDNA, is prepared with the same procedure and allows to hybridize on the glass slide.
From the millions of immobilised probes, the cDNA fragment finds its complementary sequence and hybridized with it.
The microarray scanner scans the entire slide and collects the data of hybridization. Based on the hybridization signal, the amount of gene expression can be determined.
Microarray technologies used in the transcriptomics:
- High-density oligo microarrays
- Custom microarray with fluoro-labelled cDNA
Read our article on it: Genome-On-A-Chip: DNA Microarray.
RNA- seq:
The RNA sequencing is a high-throughput method which quantifies the RNA present in a sample and also captures the sequence data.
The RNA is first reverse-transcribed into the cDNA and fragment library is constructed using the restriction digestion method.
For here, in RNA sequencing typically 100 to 1000 or10,000bp fragments are created and allow for sequencing.
The mechanism of sequencing is the same as the DNA sequence. The fragments are read in the machines and aligned based on the sequence information available with us. The “contings” are generated, aligned and computationally analysed.
RNA-seq used for the transcriptomics analysis:
- SAGE- serial analysis of gene expression
- CAGE- cap analysis of gene expression
- EST-based Sanger sequencing of cDNA
- MPSS- massively parallel signature sequencing
RT-PCR:
The cDNA constructed into the reverse transcription method can also be quantified in the RT-PCR.
Using either hydrolysis probe chemistry or probe hybridization chemistry the nucleic acid quantification is done. The signal obtained from the fluorochrome during the amplification is reported and counted.
The real-time gene expression quantification is quite an accurate method however, the major limitation of it, is the data processing.
Only a single transcript can be quantified in each reaction. Thus for a different transcript or for entire transcriptome, we have to set up different reactions.
The RT-PCR method is a good choice for a single mRNA or transcript determination.
Related article: Real-time PCR: Principle, Procedure, Advantages, Limitations and Applications.
Comparison between microarray vs RNA seq:
In both techniques, we must require mRNA as starting material, henceforth, mRNA extraction is pioneer step in both, however, for the microarray analysis the requirement of the starting material (mRNA) is higher- approximately 1μg mRNA.
Whereas for RNA-seq we need as low as 1ng of total RNA or mRNA.
The microarray method is higher throughput method as compared to RNA-seq.
Still, the microarray is low labour intensive method which relies mainly on the instrumentation and microarray set up.
On the other side, the RNA-seq method is labour intensive method required manpower for sample preparation, sequencing and data analysis.
Due to this reason, the sensitivity and dynamic range of the RNA-seq are more powerful in comparison with the microarray.
Interestingly, the reproducibility of both the method is higher.
The microarray can detect only splice variants based on the probe used in the array whereas the RNA-seq can detection splice variants as well as other variations such as SNPs.
One of the major limitations of the microarray is the requirement of the prior sequence knowledge for construcing the array.
Contrary to this, no prior sequence information is required for RNA-seq, furthermore, it can also detect new mutations.
The quantification accuracy of both the method is higher.
Transcriptome data analysis:
One of the tedious jobs is to analyse the data collected from the transcriptome analysis because it’s a huge data file, trust me!
A single transcriptome file may range from the 700mb to 2GB of raw data.
Further, previous sequence information is required to construct the probes as well as to analyse the transcriptomes data and results. Computational software and algorithms are required to read the results of RNA sequencing and microarray.
In the very first step, the data or the results are compared with the reference sequence or transcriptomes available with us.
In the RNA-seq, once the data are collected, the fragments or “contings” are first aligned and splice variants are constructed.
Then in the next step, the data or the sequence is compared with the reference sequence.
In the microarray analysis, once the image is captured, the scanner scans the amount of fluorescence emitted during the hybridization.
The final results are sent to the bioinformatics lab for comparing data with the reference data set and for interpreting results.
The objective of all of this is to understand which transcripts or RNA or mRNA are constructed and which are not.
However, sophisticated bioinformatics lab set up and experts are required to process the transcriptomics data.
Single-cell transcriptomics:
A transcriptome or gene expression analysis done from a single cell is called single-cell transcriptomics.
The purpose of the single-cell transcriptomics is to investigate gene expression from only a single cell.
For isolation of a single cell, different techniques such as cytoplasmic aspiration and micropipette are employed to separate a cell.
The mRNA is then isolated and processed through RT-PCR, droplet PCR or single-cell RNA-sequencing.
Applications of transcriptomics:
The transcriptomics is now in its growing phase, thus we do not know more about how to use it in different fields, nonetheless, several important application of it are enlisted here:
Plant breeding and hybridization.
Stem cell and cancer research.
Embryogenesis and in vitro fertilization studies.
Tissue specific gene expression studies.
Characterization of non-coding RNAs.
Retrotransposons like TEs are incorporated into the genome through reverse transcription, active transposons can disrupt gene functions and causes mutations or epigenetic alterations.
Transcriptomics like technologies is utilised to detect the presence of transposable elements in a genome.
RNA-seq like transcriptomics technologies are used in the detection of pathogenic and virulent strains present in a sample.
Conclusion:
Analysis of gene expression is one of the key goals of the transcriptomics, moreover, quantification of other non-coding RNAs can be done. This information helps scientists to understand why some of the mRNAs expressed and why some are not.
Although, data interpretation for transcriptomics is a tedious task and required a lot of expertise to perform it.
Sources:
Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T. Transcriptomics technologies. PLoS Comput Biol. 2017;13(5):e1005457. Published 2017 May 18.

This is an excellent review and very well explained.