
“Real-time PCR is a technique used for the quantification of nucleic acid present in the sample using either fluorescence dye or probe-based technique.”
PCR is capable enough to amplify the target or template DNA. Meaning, it can produce copies of DNA. It allows us to study the target sequence or a gene and therefore has been a crucial technique in genetics.
However, the conventional PCR is only capable enough to amplify the DNA. But what if we would like to quantify the DNA or even RNA? For example, what if we wish to study gene expression, viral or bacterial load or investigate food or environmental samples?
That’s not possible with just an amplification! We need to have an additional setup in our thermal cycler that can monitor the amplification and count every possible nucleic acid present in the sample.
An advanced version of the PCR which we know as a real-time PCR can perform quantitative analysis. We have covered many articles about PCR starting from its basic concept to how to optimize annealing temperature. But never talked about real-time quantification.
So you may wonder how it works. How does quantification occur in a real-time PCR, and what additional components are required? What is the principle behind real-time quantification?
Let’s find out.
In this article, we will learn about the basic concept of real-time PCR, its principle, working, steps, procedure, advantages, limitations, applications and results & analysis.
I will try to explain everything with simple explanations using either text or pictures whenever required. This article is huge! So take a cup of coffee and enjoy this article.
Before starting this article:
Common short forms:
| Abbreviation | Full name |
| PCR | Polymerase chain reaction |
| qPCR | Quantitative PCR |
| rt PCR | Real-time PCR |
| RT- PCR | Reverse transcriptase PCR |
| FRET | Fluorescent resonance energy transport |
| Taq Man | Taq DNA polymerase (Pac) Man |
Key Topics:
What is Real-Time PCR?
The name itself suggests that the real-time PCR performs analysis in ‘real-time.’ Meaning, during/ after the reaction!
What does it mean?
The machine reads each template DNA amplified in the reaction, specifically, during the reaction progression. Once the extension completes, the machine counts every amplicon, simultaneously.
Now you may wonder how it happens.
So, basically, all the reaction components will remain the same, like the conventional PCR but either an additional fluorescence dye or labeled probe is added for quantification.
Whether a probe or dye, during the positive amplification, releases a signal which the detector present in the machine, detects. So that’s a basic concept of real-time PCR.
We will discuss the principle of real-time PCR. but to make the point more clear, this question should be answered first.
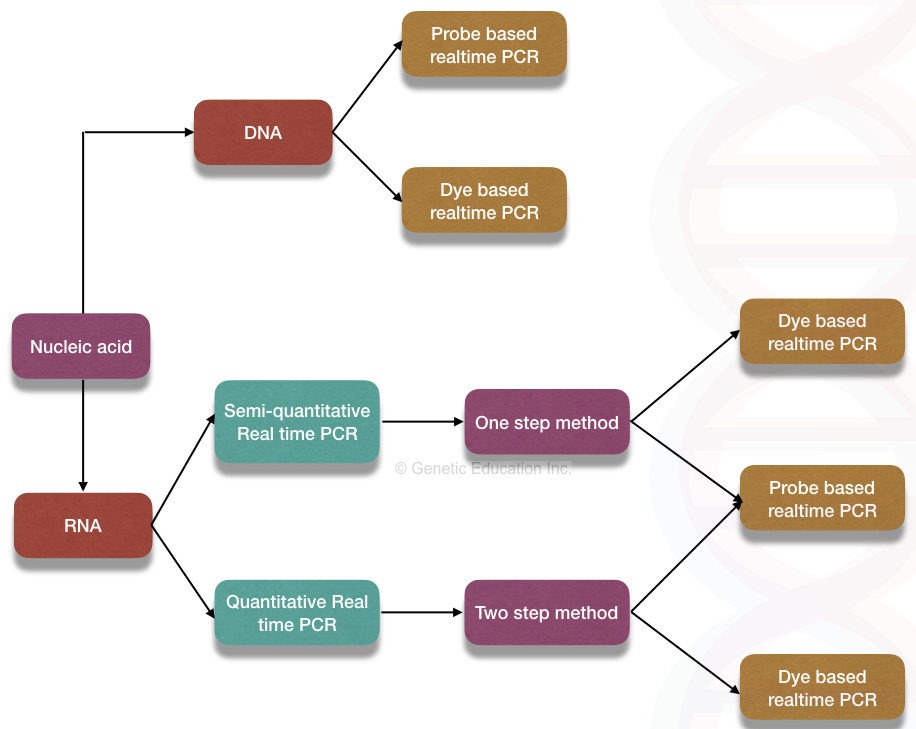
Why is the real-time PCR more advanced than conventional PCR?
The first basic reason is that the conventional PCR can’t quantify the nucleic acid present in the sample, whereas the real-time PCR can amplify and quantify the nucleic acid, simultaneously in a single reaction.
This is the reaction, real-time PCR is also known as quantitative PCR or qPCR.
The second most notable reason is downstream processing. Conventional PCR requires extensive post-processing, like– agarose gel electrophoresis for analysis. Contrary, the rt-PCR doesn’t require such analysis.
So rt-PCR is quick, easy and accurate. In addition, it also gives us the choice to select a reaction. Meaning, the reaction when doing well, can only be counted– Only a well-performing reaction counts. This minimizes the chances of error. Isn’t it amazing!
>>To learn more about this topic, read this article: PCR vs qPCR.
Common real-time PCR terminologies:
| Ct value | The point at which the fluorescent is measurable. |
| Baseline | Point of the initial amplification where the fluorescent is nearly zero. No template zone. |
| Threshold | A threshold is a set of single which distinguish amplification signals from the background signals |
| Background signals | Signals of unamplified DNA |
| Exponential phase | The phase at which the reported amplification is at its highest peak. |
Principle:
The principle of real-time PCR states that the amplification and quantification of the target nucleic acid occurs in a single reaction. Either fluorescent dye or a labeled probe is used to monitor the reaction. The more the DNA is amplified, the more fluorescence is released and detected using the fluorescence detector present in the machine.
Real-time quantification can be achieved using either DNA binding dye (intercalating dye-based method) or labeled probe (hybridization probe-based method).
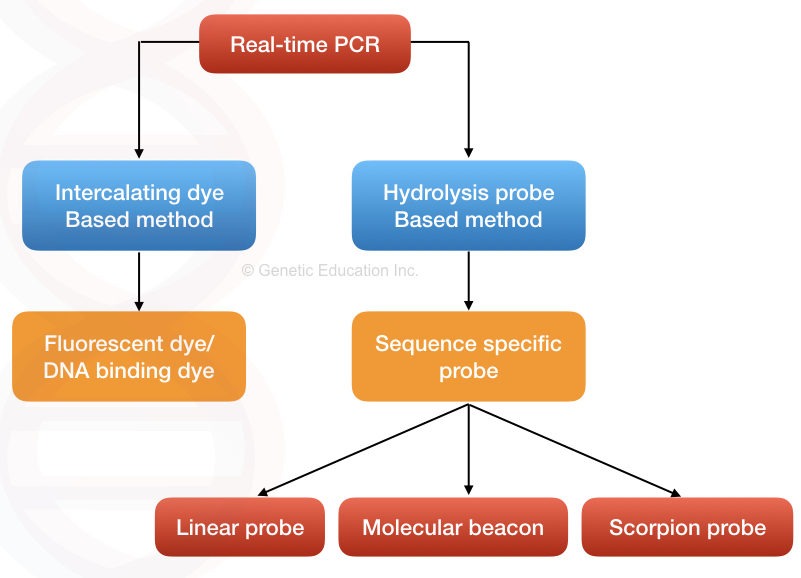
DNA binding dye-based method
DNA binding dye, also known as the intercalating dye-based method is a simple and fundamental approach for the detection, even newbies or budding scientists can perform the quantification.
The dye, either EtBr, SYBR green or any other fluorescence dye can only bind to the double-stranded DNA and emit fluorescence. So the detector detects signals only when the amplification occurs– when dsDNA is formed.
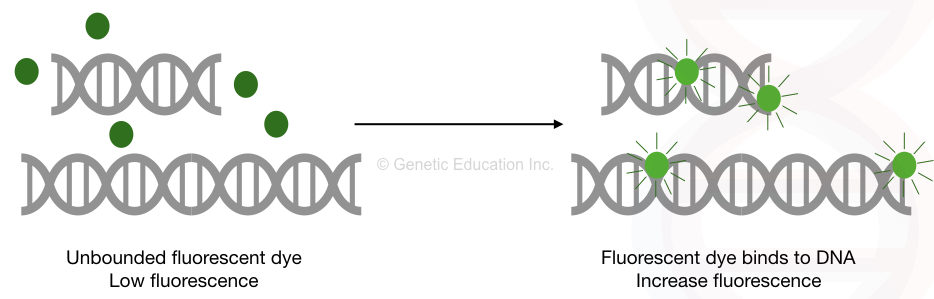
The dye has its own fluorescence. Once the dye binds to the double-stranded DNA the fluorescence emission increases 100 to 1000 fold than the original signal. However, the original dye fluorescence is taken as the baseline (as a reference) for the detection.
The result of the experiment depends on the specificity of the primers used in the PCR reaction. In case of non-specific binding or amplification, the dye will also bind to the non-specific dsDNA and emit fluorescence.
It can even give us a positive signal in the case of primer-dimers as well. Thus, the technique is less sensitive compared to the probe-based technique. SYBR green is the most common and widely used dye in real-time PCR.
Despite having limited sensitivity the present technique can be used in the detection of sensitive templates using the melting curve analysis. Melting curve analysis is explained in the results and interpretation section.
Put simply, The dye-based method is rapid, quick, reliable and cost-effective. Also, the chance of error in the experiments is less and the reaction setup is simple & easy to use.
Hydrolysis probe-based method:
In the probe-based hydrolysis method, a single-stranded fluorescent-labeled DNA probe has been incorporated into the reaction. The probe binds on the complementary location, hydrolyzes and emits fluorescence by the action of Taq DNA polymerase.
So it performs quantification in a real-time reaction during the reaction progression. This makes the technique more accurate and reliable.
Do you know?
qPCR greatly relies on the use of the TaqMan probe. “Taq” is taken from the Taq DNA polymerase while the “Man” is taken from the PacMan game. Yes, the name is actually taken from the video game, remember what the PackMan do?
Now, depending on the requirements various types of probes are used in the real-time PCR reaction.
Linear probe:
Linear probes are the TaqMan probe, which relies on the activity of Taq DNA polymerase, as we said. The probes structurally consist of labeled short single-stranded and sequence-specific DNA molecules that are radio or fluorescent-labeled.
Here the probe is labeled with the fluorescent dye described as a reporter molecule, situated at the 3’ end. The other 5’ end has the quencher dye which is in close proximity to the reporter dye and quenches the fluorescence of the reporter dye.
Now, in the probe-based method, not only the probe but the Taq DNA polymerase plays an important role. The Taq DNA polymerase used in real-time PCR has 5’ to 3’ exonuclease activity, which removes the probe by extending the DNA. Once the probe dissociates the reporter molecules emit fluorescent light.
Note: The amount of fluorescence released during each run is directly proportional to the amount of DNA amplified during the reaction.
The main advantage of the probe-based method is that we can use multiple probes for multiple template DNA sequences. This means we can amplify multiple templates in a single reaction efficiently.
The graphical representation of the probe-based hybridization method is explained in the figure below.
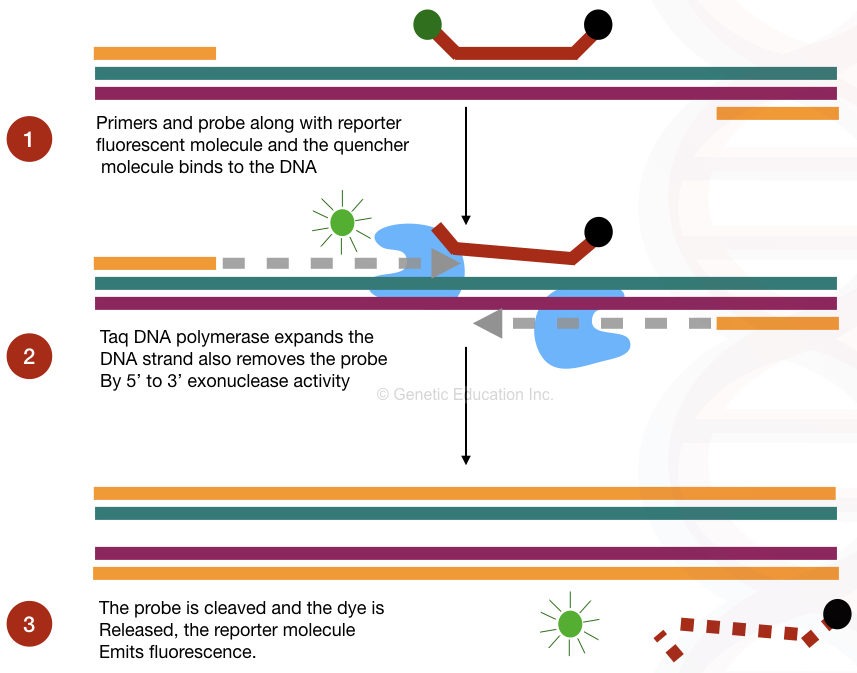
On the technical side, TAMRA and Black Hole Quencher are two widely used quencher dyes. While FAM is the most popular reporter dye. For effective probe-based detection, annealing and extension can be combined in a single reaction step, at 60ºC.
I want to discuss more about the TaqMan probe, but that’s not our article’s main focus. We will discuss the TaqMan probe in some other article and will notify it here, later.
>> Related article: DNA probes: Labeling, Types, Applications and Limitations.
So we can say in the probe-based method,
“After every round of amplification, more probes are hydrolyzed, more fluorescence is emitted and more amplicons are quantified.”
Molecular beacons:
It’s clear now that linear probes are more prone to non-specific detection, much like the dye-based technique, however, using specially designed synthetic probes known as molecular beacon probes, the problem can be solved.
So, the molecular beacon probes have been used to prevent non-specific binding and detection. Before understanding a molecular beacon, understand its structure first.
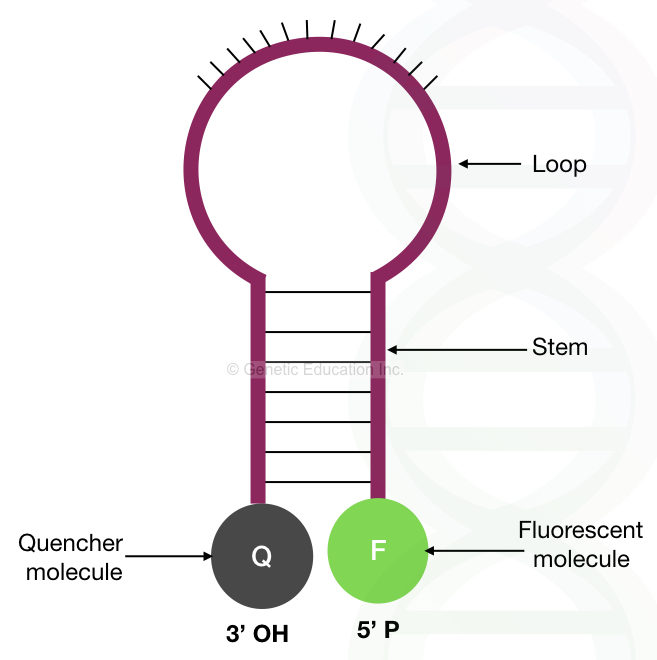
The molecular beacons rely on the mechanism of thermodynamics in which a (DNA) molecule remains in such a condition that the majority of its energy can be saved. Here instead of binding non-specifically, the molecular beacon remains in a hairpin structure.
Structurally, the complementary sequences present on both ends of the hairpin loop-like structure help to prevent non-specificity. On the other hand, the central loop is complementary to the target sequences. One end of the hairpin loop has the quencher dye and one end has the reporter fluorescent dye.
Here, interestingly, when the two ends of the hairpin stem are in close proximity to each other, the reporter molecule remains quenched and cannot produce fluorescence. However, when it binds to the complementary sequence, the two ends of the hairpin separate from each other and the reporter dye releases a fluorescent signal. The deterctor reports it as a positive amplification.
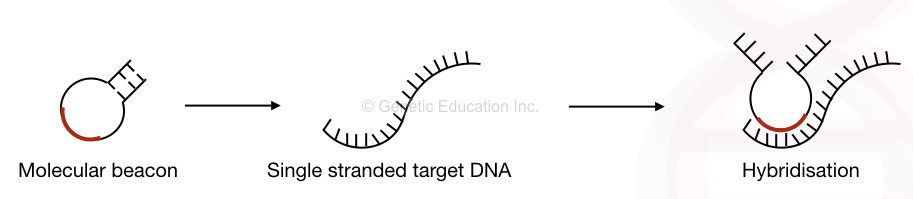
Put simply, the molecular beacon probes remain unavailable for any sequence until they find their exact complementary location. So If the probe (molecular beacon) cannot find its complementary sequence, it remains in the hairpin loop and prevents non-specific bindings.
The molecular beacon probes are highly sequence-specific and are the best choice for sensitive and low-abundant templates. It can’t compromise the sensitivity of the PCR as the mechanism does not rely on the activity of Taq DNA polymerase.
The annealing and extension occur in two separate steps at two different temperatures. Therefore, Taq Pol works at its full activity at 72°C. Nonetheless, the loop and stem designing for beacons is a crucial step in this process.
So, the success rate depends on the stem and loop structure of the molecular beacon hairpin. Too stable hairpin structure fails the reaction because it can’t hybridize and unquench while too unstable hairpin or beacon structure is useless.
Furthermore, melting curve analysis is necessary to assist the function of molecular beacons. I have written an amazing article on this topic. You can read it here:
>> Molecular Beacon: A hairpin that enhances real-time PCR specificity.
Scorpion probes:
Scorpion probes are other types of probes or we say, a type of molecular beacon used in probe-based real-time quantification.
Here, instead of a separate probe and primer, the hairpin loop is incorporated directly at the 5′ end of the primer. The 3′ end contains the complementary sequence to our target DNA. The scorpion probe is even more specific than the molecular beacons.
Keynote:
Which technique to use in the experiment (from dye or probe-based method) depends on,
- Experiment type.
- Cost or budget of the experiment.
- Knowledge and expertise of the scientist.
- Sample type.
Steps in Real-time PCR:
- Sample preparation: A high-quality and quantity DNA sample or cDNA is prepared.
- Primer and probe design: Sequence-specific primers and probes are designed using computational software or tools.
- Reaction preparation: A PCR reaction is prepared using all the standard reaction preparation parameters.
- Reaction setup: Cyclic conditions and temperature for denaturation, annealing and extension have been set up in the instrument.
- Detection: Fluorescence detection has been carried out either at the end of the reaction and during amplification.
- Data collection: The instrument records fluorescence after each round of amplification and plots the values in a graph.
- Ct value determination: The Ct value of the experiment is determined.
- Data analysis: By preparing the graph and analyzing the data, computer software provides the quantification data.
Ingredients of Real-time PCR Reaction:
The real-time PCR reaction contains all the ingredients that are used in the conventional PCR, but an additional dye or probe has been added for the quantification. Here is the list of ingredients, their function and links for dedicated articles are given below.
| Ingredient | Function |
| dNTPs | dNTPs are added during the synthesis of DNA. |
| Taq DNA polymerase | Catalyzes the amplification reaction and adds nucleotides to the DNA. |
| Template DNA | Is used as a template region for primers and probes to anneal and Taq DNA polymerase to synthesize the new strand. |
| Primers | Provide a free 3’-OH end for Taq DNA polymerase. |
| Nuclease-free water | Balances the reaction. |
| Reaction buffer | Enhance the reaction for effective amplification. |
| MgCl2 | MgCl2 is added to improve the reaction specificity. |
| Probe | Binds to template DNA and allows quantitative analysis. |
| Fluorescence dye | Binds to the dsDNA and allows quantitative analysis. |
Note: It’s important to use Hot Start DNA polymerase for effective quantification.
Results and analysis:
The crucial part of real-time PCR is results, data collection and analysis. Here, we need expertise to evaluate the results. In this section, I will explain several types of analysis that is commonly performed during real-time analysis.
Melting curve analysis:
Melting curve analysis is a type of end-point detection that quantifies templates after the completion of the reaction. This type of analysis is commonly used for dye-based real-time PCR for accurate quantification.
So after the completion of the reaction, the temperature is gradually increased to melt the double-stranded DNA. Meaning, the copies of the dsDNA generated are now melted or separated.
Eventually, the dye dissociates from the dsDNA and the fluorescence signals gradually decrease. A single and sharp peak for all of the templates in the graph indicates specific amplification, while the rest of the peaks are either non-specific products or primer dimers.
We can say,
“A larger sequence needs more time and higher temperature for melting while non-specific amplicons need the lower and varied temperature to melt and so give shorter curves in a graph.”
I have tried putting this explanation as a graph, you can see the fluorescence vs. melting temperature graph below,
The fluorescence vs melting temperature graph is also called a dissociation curve and the method is called a dissociation curve analysis.
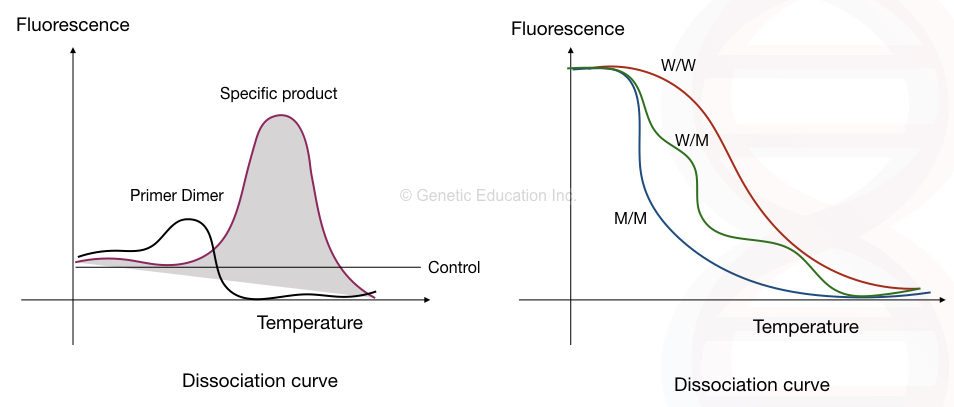
The first image shows the presence of primer-dimer while the second image shows the dissociation curve of different alleles at different temperatures.
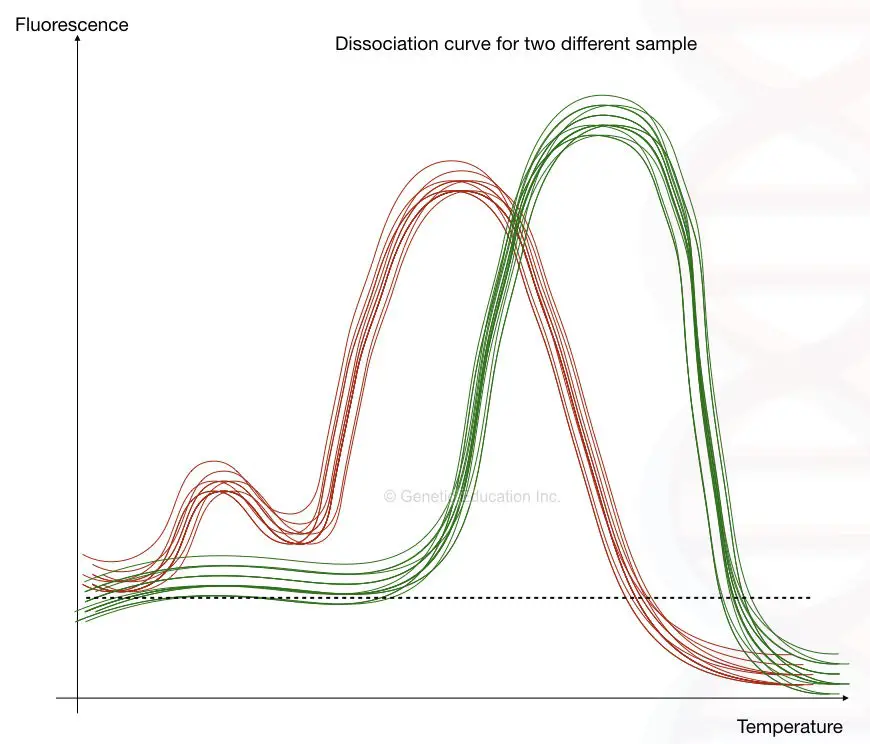
Standard curve analysis:
Standard curve or real-time analysis is entirely different from melting curve analysis or endpoint PCR. Here, quantification occurs during the reaction progression, especially during the exponential phase.
In the standard curve analysis method, the serially diluted sample or template is quantified against the known template.
Here the known template is serially diluted many times and quantified. The source of the information is used for the sample and unknown template which is also serially diluted and measured against the known.
In simple words, we can say that each unknown sample dilution is compared with each known standard dilution.
The method is also referred as absolute quantification. The method is one of the best choices for the viral and bacterial load quantification. Also, the absolute quantification method is rapid and more accurate.
By comparing the Ct value of both the standard and the unknown template, the linear curve graph is generated.
For each known and unknown dilution, the Ct of all is plotted on the graph and by comparing the data the initial concentration of the unknown template is determined.
However, the method of calculation contains so much maths, hence we are not discussing it but it is automatically calculated by the machine.
Relative quantification:
Another method is for those types of templates which do not have reference values. Or it is totally unknown.
Here, the calibrator is used to create the baseline for the experiment, and with respect to the baseline calibrate and the Ct value of the template sample, and the amount of the expression of a gene into the unknown sample can be determined.
The figure below shows the graph of different templates,
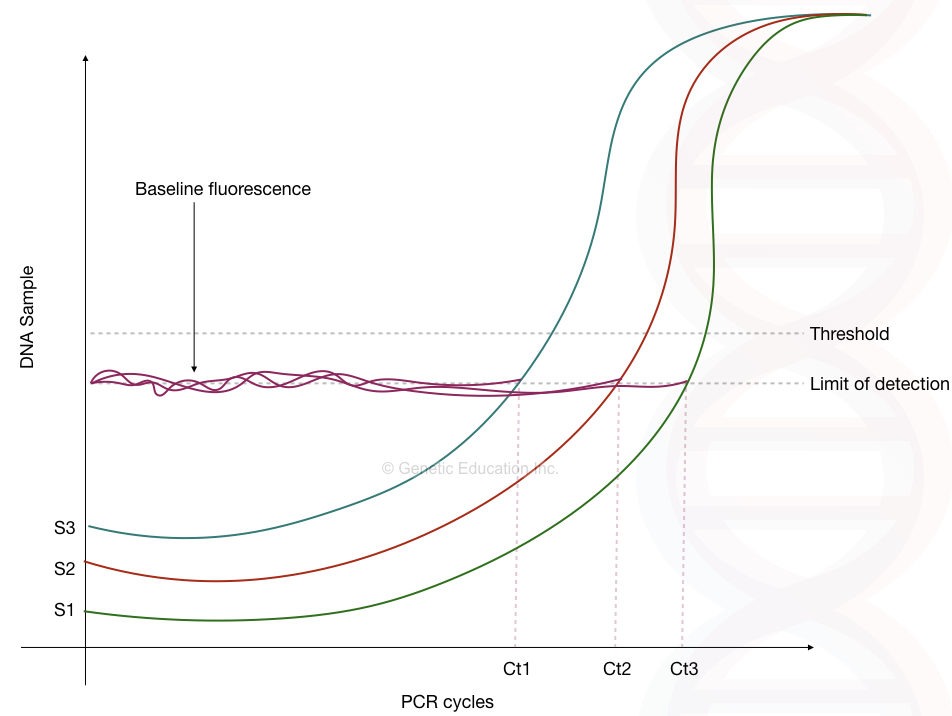
Absolute quantification:
In this type of quantification, a reference gene or sample is available for performing the quantification. Absolute quantification is more accurate.
Advantages of Real-time PCR:
The method is cost-effective.
The conventional PCR method is more costly than the qPCR due to the use of many other chemicals and agarose gel electrophoresis.
It is time-efficient.
Definitely, it is.
The average time consumed by the PCR reaction along with the agarose gel electrophoresis and data interpretation is approximately 4 to 4.5 hours.
Conversely, real-time qPCR results in ultra-fast time. The average duration of the qPCR reaction is around 30 minutes to 2 hours.
More sensitivity and specificity.
The quantitative real-time PCR method is more sensitive, specific and efficient.
Though the probes and primers are highly sequence-specific, if any nonspecific bindings occur, it is monitored immediately during the reaction. Also, the main reaction or the quantification of our template cannot be influenced by the non-specific bindings.
Fewer templates required:
The overall assay required less amount of the template material. It required 1000-fold less template DNA or RNA for the reaction compared with the conventional PCR.
Melting curve analysis:
The main advantage of quantitative PCR is the amplicon validation through the melting curve analysis.
We can quantify how many amplicons are generated and how many non-specific or primer-dimers are formed during the PCR reaction by doing the melting curve analysis.
The major advantage over the other PCR techniques is the quantification. It quantifies the template DNA or RNA present in the sample.
Only Real-time is sufficient:
No post-PCR and data processing is required in the quantitative real-time PCR. As with conventional PCR, agarose gel electrophoresis gel-based interpretation is not needed in the qPCR.
Limitation of real-time qPCR
Although the advantages of quantitative rt-PCR are far more than the conventional PCR, still the technology has several limitations.
- The instrument itself is too costly compared with conventional PCR.
- Also, the multiplexing is still limited in Real-time PCR.
- Kits are not available for all kinds of genes and disorders.
- Fewer standard protocols are available.
- Expertise and experience are required to perform the real-time analysis and evaluate the data.
Applications of quantitative PCR:
Although the main application of real-time PCR is in gene expression studies, it’s nowadays used in various fields for various purposes. Here are some examples of some of the crucial applications of real-time PCR.
Disease diagnosis:
One of the most powerful uses of any PCR technique is in inherited disease diagnosis.
The real-time PCR is used in the diagnosis of single-gene and multi-gene diseases. It is used to quantify the mutated gene in the disease patient. Quantitative real-time PCR is even used in the determination of copy number variation in different tissues for different inherited disorders.
microRNA analysis:
MicroRNAs are smaller RNA molecules of 20 to 25 nucleotides in length. It plays an important role in the gene regulation pathway. The qPCR assay is used to quantify the microRNA from different tissues.
By quantifying it we can estimate the gene expression level in different tissues influenced by the microRNA.
Cancer detection and studies:
Circulating tumor cells contain the mutant mRNA and spread to different tissues if it is malignant. By quantifying the total mRNA against the mutant mRNA in the sample, the cancer stage can be predicted. Cancer severity can also be estimated in this way.
Note that, the mutant mRNA is the best biomarker for gene expression studies for cancer.
Furthermore, therapy or drug response can also be determined quantitatively using before and after-treatment gene expression analysis. Thus, it can effectively estimate treatment, therapy or drug success rate.
In conclusion, real-time PCR is useful for cancer detection, diagnosis, prognosis and monitoring for treatment response.
Microbial load testing:
Real-time PCR allows us to conduct accurate microbial load quantification from various biological samples including disease samples, soil, water, food or environmental samples, microbial cultures or any other sample containing microbes.
In addition, It can also help to estimate the active microbial load from the sample and help in microbial risk assessment.
GMO detection:
Genetically modified organisms are organisms whose genetic makeup is altered using genetic engineering or transgenic techniques specifically in plants, animals and microorganisms.
Now, this is something interesting,
Inserting DNA through the vector is not sufficient to do so. For the successful development of GMOs, one has to quantify the expression of the protein formed by the inserted gene.
For that, only insert validation isn’t sufficient. We need to study the expression or mRNA to understand the efficacy of the insert. Real-time PCR helps determine accurate gene expression quantification for the insert.
Thus, it helps in not only insert validation but also in estimating the insert success rate.
Genotyping and quantification of pathogens:
Microbial infections are the second most common reason for worldwide mortality and morbidity. Unlike conventional PCR (which can only detect a single or a few strains and genotyping), real-time PCR is used to determine the infectivity or genotypes of many different strains.
It can actually determine the microbial load present in the infection sample and give us accurate data about the infection, infection stage and drug response.
Melting curve or dissociation curve analysis and Ct value analysis help in investigating the severity of the infection. The most recent examples are the detection and quantification of COVID-19 coronavirus and H1N1 swine flu.
q(RT)- PCR helps in both pandemics which quantify the absolute amount of virus present in a sample in quick time. This article will help you to learn more: What is the Ct value of SARS-CoV-2 (COVID-19) RT-PCR?
Moreover, the qPCR is applicable in the Identification, characterization, genotyping and quantification of an infectious pathogen.
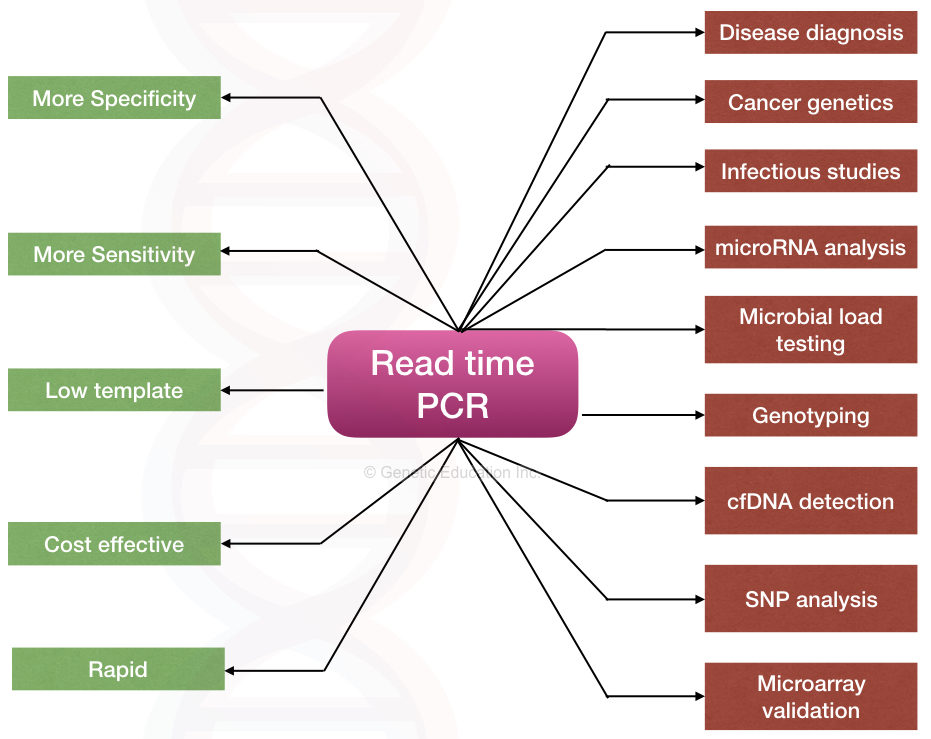
Identification and quantification of circulating nucleic acid:
Cell-free fetal DNA and circulating microRNA are two types of circulating nucleic acid present in the bloodstream. Both types of nucleic acids are present in very little quantity.
Even if it is very difficult to extract cell-free DNA, qPCR helps in the identification and quantification of circulating nucleic acid. Circulating tumor DNA, which is a type of cell-free DNA, is an excellent biomarker for cancer studies.
Any type of cell-free DNA can be quantified using the real-time PCR.
Detection of pathogenic SNPs:
Pathogenic SNPs can be identified and quantified using real-time melting curve analysis. Here, a small amount of mutant or pathogenic allele can be quantified from the sample (low abundant nucleic acid).
This helps in studying the disease stage, treatment options and monitoring the disease progression.
Apart from these applications, real-time PCR is also used in forensic studies, evolutionary studies, mutation creation, fossil studies and other applied fields.
Read more:
Wrapping up:
Real-time PCR proved to be an excellent weapon during the recent COVID-19 pandemic. It allows rapid, accurate and sensitive detection of nucleic acid present in the sample and thus is a valuable tool.
Notedly, it has been widely used in the field of epigenetic studies and is still utilized. Both dye-based and probe-based methods have their own advantages and limitations and the selection depends on the requirement of the experiment.
Learning real-time PCR will add an amazing skill set to your pocket as a scientist, so you should have to learn this technique to uplift your scientific career. I hope you like this article. Please share it and bookmark the page for future reference.
Sources:
Robert E (2010). RT-PCR: A Science and Art form. RNA methodologies 4th Edition, Academic Press, 385-448.

very detailed and descriptive information thank you so much
Thank you so much sweety
Very informative! Keep up the good work !
Good piece of information. Thanks
Thank you Jayantha
Nice article
Very nice information
Thank you Vijay Verma
sources ?