A person suffers from infertility and visits a fertility clinic for an assessment.
Everything looked perfectly normal until the doctor requested a karyotype analysis.
Shockingly, the results revealed something the patient had never expected.
To understand what was happening, the medical team performed additional investigations using qPCR and FISH (Fluorescence In Situ Hybridization). The findings were even more surprising and explained why the patient had been struggling with infertility for years.
Today’s case study is about one of the rarest genetic conditions on Earth: 46,XX male sex reversal syndrome, a condition where a genetically female chromosome complement develops into a phenotypic male.
Sounds fascinating, let’s explore it!
Key Topics:
Key Clinical Information
46,XX male sex reversal syndrome, also known as de la Chapelle syndrome, is an extremely rare disorder of sex development in which an individual has two X chromosomes and completely lacks a Y chromosome, but still develops as a male.
The condition has an estimated prevalence of 1 in 20,000–25,000 male births worldwide.
There are two major forms of this condition:
- 46,XX SRY-positive, where the SRY gene is present despite the absence of the Y chromosome.
- 46,XX SRY-negative, where the SRY gene is absent.
The SRY-positive form accounts for nearly 90% of reported cases. These individuals usually have a normal male appearance and develop typical secondary sexual characteristics. Because of this, the condition often goes unnoticed until adulthood, when infertility becomes the primary reason for medical evaluation.
Patient Summary
The patient was a 39-year-old man who presented with long-standing infertility. There was no family history of infertility or known genetic disorders.
Clinical examination revealed a normal male phenotype with well-developed secondary sexual characteristics. However, a closer examination identified small testes, mild gynecomastia, azoospermia, and reduced libido.
Hormonal evaluation showed elevated follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels, along with low total testosterone levels, indicating primary testicular failure.
Although the patient appeared completely normal externally, the underlying genetic abnormality had remained undetected for nearly four decades.
The Genetic Investigation
Kouvidi et al. (2022) reported this remarkable case while investigating the patient’s infertility.
The diagnostic workup began with conventional karyotyping, which was performed using peripheral blood lymphocyte culture.
The result was unexpected.
Instead of the expected 46,XY chromosome complement, the patient had a 46,XX karyotype, with no obvious structural chromosomal abnormalities.
At this point, the clinicians strongly suspected 46,XX male sex reversal syndrome.
However, one important question remained unanswered.
Is the SRY gene present or not?
To answer this, the researchers performed both quantitative PCR (qPCR) and Fluorescence In Situ Hybridization (FISH).
The qPCR analysis confirmed the presence of the SRY gene.
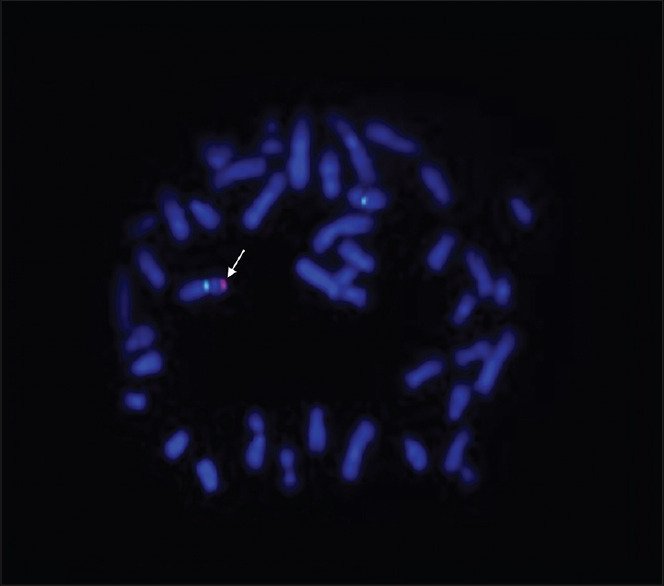
The FISH analysis provided the final piece of evidence.
The researchers used locus-specific probes for the X chromosome (green) together with an SRY-specific probe (red).
Instead of detecting the red signal on a Y chromosome, because it is not present, the SRY probe hybridized to one of the X chromosomes.
This finding confirmed that the SRY gene had translocated from the short arm of the Y chromosome (Yp) to the short arm of the X chromosome (Xp).
The diagnosis was now complete.
The patient had 46,XX SRY-positive male sex reversal syndrome.
Genetics Explained
To understand this condition, we first need to understand how sex chromosomes behave during meiosis.
Most human chromosomes exist as homologous pairs and undergo homologous recombination during prophase I of meiosis. This involves exchanging genetic material to increase genetic diversity.
The sex chromosomes are different.
The X chromosome and Y chromosome are not true homologs. Nearly 95% of the Y chromosome does not recombine with the X chromosome.
Only two small regions at the tips of the chromosomes, known as the pseudoautosomal regions (PAR1 and PAR2), participate in recombination.
These PAR regions allow the X and Y chromosomes to pair correctly during meiosis.
Occasionally, recombination extends beyond the normal PAR boundary.
When this happens, genes located just outside the PAR, particularly the SRY (Sex-determining Region Y) gene, may accidentally be transferred from the Y chromosome onto the X chromosome.
This rare event occurs randomly during spermatogenesis and cannot be predicted or prevented.
If the entire SRY gene is successfully transferred, the embryo follows the male developmental pathway despite having two X chromosomes.
As a result, the individual develops as a phenotypic male with normal external genitalia and secondary sexual characteristics.
However, because the Y chromosome itself is absent, other critical regions required for sperm production, including the AZFa, AZFb, and AZFc regions, are also missing.
Without these regions, normal spermatogenesis cannot occur, leading to severe male infertility, most commonly azoospermia.
The condition becomes even more severe in 46,XX SRY-negative individuals, where the SRY gene is completely absent. These patients often exhibit significant abnormalities of gonadal development, ambiguous genitalia, or other disorders of sex development due to the failure of normal testicular differentiation.
Key Learning
Here are the key learnings from this case study:
- Nature is remarkably precise, but even tiny errors during meiosis can have profound biological consequences.
- Male infertility is a multifactorial condition, yet chromosomal abnormalities remain one of its most significant genetic causes.
- This case also demonstrates why conventional karyotyping should not be overlooked. A simple chromosome analysis raised the first suspicion, while molecular techniques such as qPCR and FISH confirmed the diagnosis by identifying the misplaced SRY gene.
Each technique contributed a different piece of the puzzle, and together they provided a complete genetic diagnosis.
Wrapping Up
Sex reversal syndrome remains one of the rarest and most fascinating conditions in human genetics. It reminds us that biological sex is determined not simply by the presence or absence of the Y chromosome, but by the correct location and function of key developmental genes such as SRY.
Cases like these also highlight the importance of integrating cytogenetics with molecular genetics in clinical practice.
Interestingly, I had the opportunity to be part of the team that reported one of the 46,XX male sex reversal cases from India, making this condition particularly memorable in my own journey as a cytogeneticist.
Reference: Kouvidi, Elisavet et al. “A 46,XX Karyotype in Men with Infertility: Two New Cases and Review of the Literature.” Journal of human reproductive sciences vol. 15,3 (2022): 307-317. doi:10.4103/jhrs.jhrs_100_22.