Imagine a pregnant woman undergoing NIPT. The results show a rare numerical abnormality.
Let’s say trisomy 16.
She immediately visits her genetic counselor and shows the results. Under ideal conditions, many people might assume that such a result indicates a severely affected fetus and may even think about pregnancy termination.
But instead, the genetic counselor advised another genetic test.
Why?
Is the counselor not sure about the results?
Are the results misleading?
No!
The reason is that the fetus may not actually have a trisomy. This can happen because of a rare condition during pregnancy called CPM- Confined Placental Mosaicism.
Let’s understand this fascinating condition through a real case study.
[Case study #5]: Two X Chromosomes, Still a Male? A Rare Infertility Case Explained.
Key Topics:
Key Clinical Information
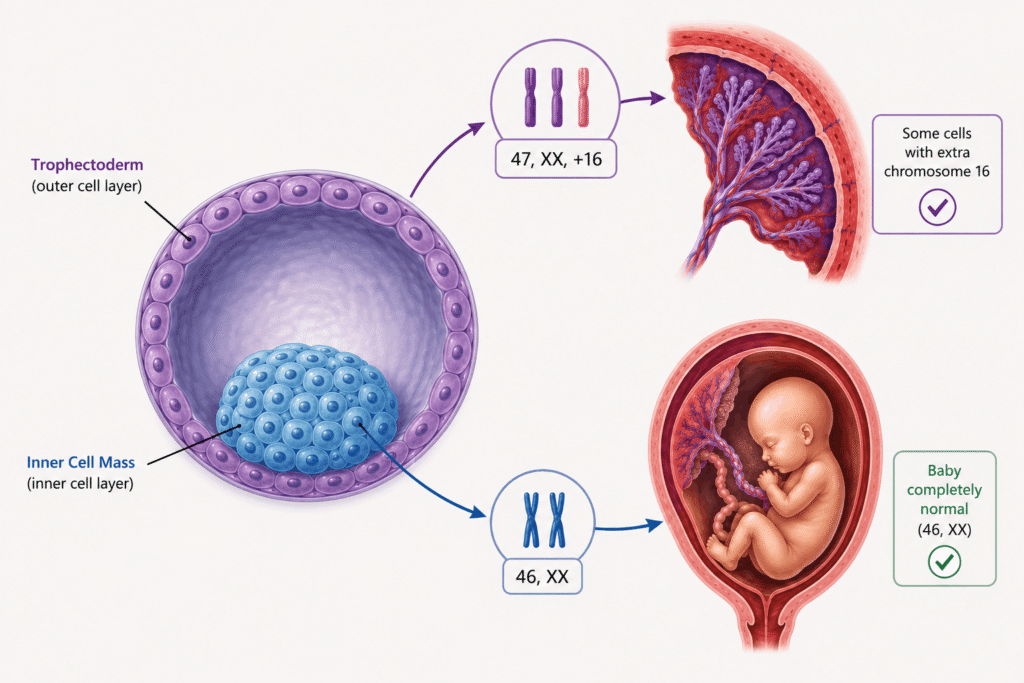
Confined Placentral Mosaicism (CPM) is a rare genetic condition, often observed during pregnancy. Here, the placenta and the fetus have different chromosomal constitutions.
For instance, the placenta may have a trisomy, monosomy or any other chromosomal abnormality, while the fetus has a completely normal chromosomal complement.
But the fetus doesn’t have any of these chromosomal abnormalities. Meaning, the fetus is completely normal.
Approximately 1-2% of pregnancies show CPM. Interestingly, CPM is one of the major biological reasons for false-positive and, less commonly, false-negative NIPT results.
The reason? Again, NIPT primarily analyzes placental DNA and not fetal DNA.
Related article: What Is Cell Free Fetal DNA Test?
Patient Summary
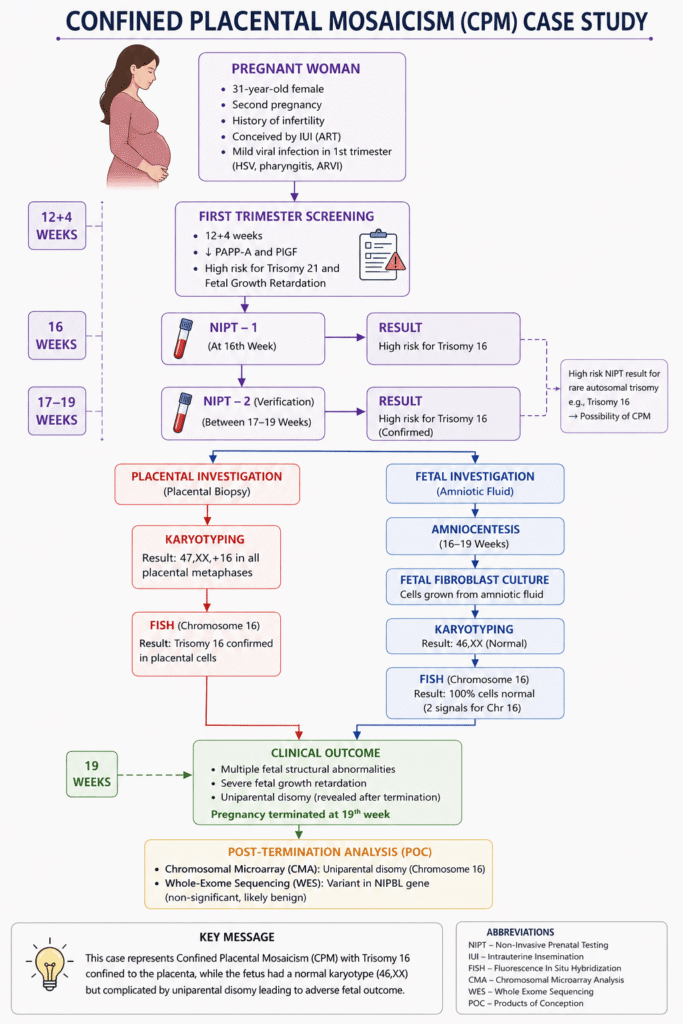
The patient was a 31-year-old woman in her second pregnancy at 16 weeks of gestation. She had a history of infertility. Her first pregnancy was achieved using assisted reproductive technology (ART) with intrauterine insemination following controlled ovarian stimulation.
The first trimester screening at 12 + 4 Weeks showed reduced PAPP-A and PIGF levels.
The screening indicated a high risk for chromosomal abnormalities and fetal growth retardation (FGR).
The mother experienced a mild viral infection during the first trimester (HSV, pharyngitis, ARVI).
Genetic Investigation
The patient underwent:
- Non-Invasive Prenatal Testing (NIPT)
- Fetal fibroblast culture
- Karyotyping
- FISH (Fluorescence In situ Hybridization)
- Chromosomal Microarray (after termination)
- Whole Exome Sequencing (after termination)
The first NIPT was performed at 16 weeks, followed by a repeated NIPT verification between 17 and 19 weeks of pregnancy.
Karyotyping and FISH were performed during pregnancy, while CMA was performed after pregnancy termination.
The initial NIPT indicated a high risk for trisomy 16, which was confirmed by the repeated NIPT, suggesting the possibility of CPM.
Karyotyping performed on the placental biopsy showed 47,XX,+16 in all analyzed placental metaphases. FISH confirmed the same finding.
To verify whether the fetus was also affected, the team collected amniotic fluid and established a fetal fibroblast culture.
Interestingly, the fetal results were completely different
Fibroblast culture revealed a normal karyotype (46,XX) without any extra copy of chromosome 16. The FISH analysis further confirmed that 100% of fetal cells carried a normal chromosomal complement.
Despite the normal fetal karyotype, multiple structural fetal abnormalities, severe fetal growth restriction, and uniparental disomy (UPD) were identified. After extensive counseling, the couple decided to terminate the pregnancy at 19 weeks.
Chromosomal microarray performed on the products of conception confirmed UPD of chromosome 16, while whole-exome sequencing identified a likely benign variant in the NIPBL gene, which was considered clinically insignificant.
Genetics Explained
This case represents a complex combination of Confined Placental Mosaicism Type III and uniparental disomy (UPD).
CPM occurs when the placental cells and fetal cells have different chromosomal constitutions, as aforementioned.
During early embryonic development, the fertilized egg divides repeatedly and eventually forms a blastocyst. The outer cell layer (trophectoderm) develops into the placenta, whereas the inner cell mass develops into the fetus.
Because these two cell lineages separate very early, a chromosomal error occurring in one lineage may not affect the other and that’s the basic foundation for CPM.
The placenta naturally contains more chromosomal abnormalities than fetal tissues because its cells divide rapidly throughout pregnancy. Since its primary role is to supply oxygen and nutrients to the fetus, it can often continue functioning despite carrying abnormal cells.
Therefore, abnormalities such as trisomy 16 may remain confined to the placenta without directly affecting the fetal chromosome complement.
However, an abnormal placenta may not function efficiently, resulting in placental insufficiency, fetal growth restriction, and poor pregnancy outcomes.
In some pregnancies, a fetus that initially carries trisomy can undergo a process known as trisomic rescue. During this process, the extra chromosome is lost from one of the daughter cells, restoring the normal chromosome number.
But here is an interesting catch!
Because the cell cannot determine which chromosome copy should be removed, both remaining chromosomes may occasionally originate from the same parent. This condition is known as uniparental disomy (UPD).
In this case, both copies of chromosome 16 originated from a single parent. Now you may have a question: Is this a problem?
Yes, it is!
Depending on the chromosome involved, UPD may disturb genomic imprinting and gene expression, leading to developmental abnormalities despite a normal chromosome count.
Key Outcomes
This case demonstrated that an abnormal NIPT result does not always indicate an abnormal fetus.
Although the placenta showed complete trisomy 16, the fetal cells had a completely normal 46,XX karyotype, confirming Confined Placental Mosaicism.
Further investigations revealed uniparental disomy of chromosome 16, explaining the adverse fetal findings despite the normal fetal chromosome count.
The case also highlights the importance of combining multiple genetic techniques before making irreversible clinical decisions such as pregnancy termination. A single screening test should never be interpreted in isolation, particularly when rare chromosomal abnormalities are detected.
Key Learnings
Pregnancy is a highly complex biological process, and conditions such as CPM demonstrate that placental genetics and fetal genetics are not always identical.
Although CPM may not directly affect the fetal chromosome complement, it can significantly impair placental function, leading to fetal growth restriction and adverse pregnancy outcomes.
NIPT is an excellent screening test but may produce false-positive or, less commonly, false-negative results because it analyzes cell-free placental DNA, not fetal DNA.
Conventional cytogenetic techniques such as karyotyping and FISH remain invaluable for investigating complex prenatal cases.
However, chromosomal microarray plays an important role in identifying uniparental disomy and other submicroscopic chromosomal abnormalities that cannot be detected by routine karyotyping.
Related Article: 6 Types of Microarray-based Genetic Testing.
Wrapping Up
NIPT has transformed prenatal screening by providing a safe, non-invasive, and highly accurate method for detecting common chromosomal abnormalities. Still, biological conditions such as Confined Placental Mosaicism remind us that no screening test is perfect.
When rare autosomal trisomies or unexpected results are detected, confirmatory testing using amniocentesis, karyotyping, FISH, and chromosomal microarray becomes essential before making critical clinical decisions.
This case also demonstrates an important principle in prenatal genetics: an abnormal placenta does not always mean an abnormal baby and genetic counseling plays a significant role in such complicated cases.
Reference:
Mykytenko, D. O. et al. (2022). Placental mosaicism: complete discordance between the placenta and the fetus. Clinical case record. Wiadomości Lekarskie, 75(3), 742–746. https://doi.org/10.36740/WLek202203130.